
Vol. 42, n.º 1, 2009
|
REVISTA
ESPAÑOLA DE
Vol. 42, n.º 1, 2009 |
CASUÍSTICA
Inmaculada Barredo Santamaría, Iñaki Zabalza Estévez, Alberto Saiz López, Igone Imaz Murga, Eduardo de Miguel Herrán, José Antonio Álvarez Martínez, Amaia Mariscal Polo1, Ibon Bilbao Badiola1
Servicio de Anatomía Patológica. Hospital
Galdakao-Usansolo. Barrio Labeaga, s/n. Galdakao. Vizcaya. España.
1 Servicio de Dermatología.
inmaculada.barredosantamaria@osakidetza.net
RESUMEN
El neurotecoma celular es una tumoración cutánea benigna poco frecuente de histogénesis incierta, que ocurre fundamentalmente en la parte superior del cuerpo de niños y adultos jóvenes, con predominio femenino. Clásicamente se considera que los neurotecomas (variantes mixoide, celular y mixta) forman parte de un espectro de tumores con supuesto origen en la vaina nerviosa. Sin embargo, la falta de diferenciación neurosustentacular convincente tanto en el neurotecoma celular como en el mixto ha cuestionado este concepto, y numerosos autores afirman que se trata de tumores diferentes que justifican una clasificación separada. Se presentan dos casos de neurotecoma celular, en un varón de 24 años y en una niña de 9 años, con tumoraciones asintomáticas en el hombro derecho y en la rodilla izquierda respectivamente. Histológicamente, ambos casos se caracterizan por una proliferación en la dermis reticular de células fusiformes y/o epitelioides dispuestas en nidos y fascículos, rodeados por un estroma colágeno denso. Uno de los casos muestra importante atipia citológica y un patrón marcadamente plexiforme. Inmunohistoquímicamente, ambos casos son positivos para vimentina, CD68, CD10, factor XIIIa y actina músculo-liso. En uno de los casos también hay inmunorreactividad para enolasa neuronal específica. El resto de los marcadores realizados son negativos, entre ellos la proteína S-100. Los hallazgos inmunohistoquímicos sugieren diferenciación fibrohistiocitaria, lo que probablemente justifica la separación del neurotecoma celular del neurotecoma mixoide. En cuanto al neurotecoma mixto, éste probablemente representa una variante mixoide del neurotecoma celular.
Palabras clave: Neurotecoma, neurotecoma celular, inmunohistoquímica, piel.
SUMMARY
Cellular neurothekeoma is a rare benign cutaneous neoplasm of uncertain histogenesis arising usually on the upper body of children and young adults, with a female predominance. Neurothekeomas (myxoid, cellular and mixed variants) are classically regarded as forming part of a spectrum of tumors of putative nerve sheath origin. The lack of convincing evidence of neurosustentacular differentiation in cellular and mixed variants, however, has challenged this concept, and numerous authors state that they are different tumors that warrant a separate classification. This is the report of two cases of cellular neurothekeoma, in a 24-year-old man and a 9-year-old girl, with asymptomatic tumors occurring on the right shoulder and left knee respectively. Histologically, both cases are characterized by a proliferation of spindle and/or epithelioid cells involving reticular dermis, arranged in fascicles and nests surrounded by sclerotic collagen. One of the two cases show significant cytologic atypia and a notably plexiform pattern. Immunohistochemically, both cases are positive for vimentin, CD68, CD10, factor XIIIa and smoth muscle actin. In one of the two cases there is also immunoreactivity for neuron-specific enolase. The remainder of immunomarkers are negative, S-100 protein included. The immunohistochemical findings suggest fibrohistiocytic differentiation, so it probably warrants the separation of cellular neurothekeoma from myxoid neurothekeoma. In respect to the mixed neurothekeoma, this probably represents a myxoid variant of cellular neurothekeoma.
Keywords: Neurothekeoma, cellular neurothekeoma, immunohistochemistry, skin.
INTRODUCCIÓN
Los neurotecomas son un grupo de neoplasias benignas, predominantemente cutáneas, que se clasifican generalmente en: 1) variante mixoide o clásica (también llamado mixoma de la vaina nerviosa), 2) variante celular, y 3) variante mixta o intermedia, que muestra hallazgos histológicos intermedios entre el neurotecoma mixoide y el celular.
Los neurotecomas mixoides fueron descritos por primera vez en 1969 por Harkin y Reed en la segunda serie de los atlas de patología tumoral del Instituto de Patología de las Fuerzas Armadas (AFIP), con la denominación de «mixoma de la vaina nerviosa» (1). Otros nombres utilizados por diferentes autores para nombrar estos tumores son: neurofibroma paciniano, neurofibroma cutáneo bizarro, neuromixoma lobulillar cutáneo y mixoma perineural.
Los neurotecomas celulares fueron descritos en 1980 por Gallager y Helwig, quienes acuñaron el término «neurotecoma» (del griego qeke: vaina) para acentuar su posible origen en la vaina nerviosa (2). Rosati y cols. describieron en 1986 una lesión similar a la que denominaron «neurotecoma celular» (3).
Además de las variantes mixoide y celular del neurotecoma, se introdujo posteriormente un tercer subtipo con características híbridas, denominado neurotecoma «mixto» o «intermedio» (4). La descripción de estas lesiones «mixtas» favoreció el concepto de que las variantes mixoide y celular eran los dos polos de un espectro morfológico de tumores con origen en la vaina nerviosa. Sin embargo, la falta de diferenciación neurosustentacular convincente tanto en el neurotecoma celular como en el mixto ha cuestionado este concepto, y numerosos autores señalan que se trata de tumores diferentes que justifican una clasificación separada (5-9).
Existe bastante confusión en la terminología. De hecho, el grupo del AFIP segrega definitivamente el mixoma de la vaina nerviosa del grupo del neurotecoma, manteniendo la denominación «neurotecoma» para el resto, es decir para los denominados neurotecoma celular y mixto (6,10). Para complicar más las cosas, dependiendo de la cantidad de matriz mixoide, los autores subclasifican los neurotecomas en celular, mixto y mixoide (6).
Se describen las características clínicas, histológicas e inmunohistoquímicas de dos casos de neurotecoma celular.
DESCRIPCIÓN DE LOS CASOS
Caso 1
Varón de 24 años, sin antecedentes de interés, que presenta un nódulo cutáneo eritematoso y lobulado en el hombro derecho, asintomático, de 2 años de evolución. Se extirpa con el diagnóstico clínico de quiste epidérmico, queloide u otros. En el examen macroscópico se identifica una cuña cutánea con un nódulo elevado de 0,7 cm de diámetro. Tras la sección por el eje mayor se incluye totalmente para su estudio histológico convencional.
Microscópicamente se observa una tumoración dérmica mal delimitada con patrón de crecimiento plexiforme (fig. 1). Está constituida por numerosos nódulos redondeados y ovalados de tamaño variable, algunos de aspecto arremolinado, así como por fascículos cortos y algunas áreas sólidas (fig. 2). Las células tumorales tienen morfología fusiforme y epitelioide, un núcleo ovalado o elongado, vesiculoso, con pequeño nucléolo, y un citoplasma ligeramente eosinófilo, finamente vacuolado (fig. 3). Con frecuencia se observan núcleos agrandados, de contorno irregular, con presencia de multilobulación y multinucleación (fig. 4). La tasa de mitosis es de 3 por 10 campos de gran aumento. No se evidencian fenómenos de necrosis. El estroma es colágeno denso, y muestra un moderado infiltrado inflamatorio linfocitario, que penetra entre las células tumorales. Se observa una moderada cantidad de mucina azul-alcián positiva, fundamentalmente en los nidos tumorales de menor tamaño. El tumor respeta la porción más profunda de la dermis reticular y deja una zona fina de dermis superficial no afectada. La epidermis está aplanada.
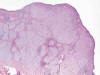
Fig. 1:
Tumoración dérmica con patrón de crecimiento plexiforme.
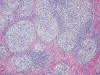
Fig. 2:
Nódulos tumorales rodeados por un estroma colágeno denso.
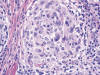
Fig. 3:
Células tumorales fusiformes y epitelioides, con citoplasma ligeramente
eosinófilo y núcleo vesiculoso con pequeño nucléolo.
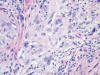
Fig. 4:
Células con núcleos grandes, multilobulados, y células tumorales multinucleadas.
Las técnicas de inmunohistoquímica muestran marcada positividad en las células tumorales para vimentina, CD68 (fig. 5) y CD10 (fig. 6), y ligera positividad para factor XIIIa y actina músculo-liso. Son negativos: S-100 (fig. 7), HMB-45, AE1/AE3, EMA, CD34, CD21, CD35, PGFA, enolasa neuronal específica, sinaptofisina, cromogranina, desmina, calponina y colágeno IV.
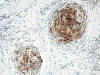
Fig. 5:
Inmunorreactividad para CD68.
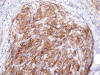
Fig. 6:
Inmunorreactividad para CD10.
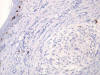
Fig. 7:
Ausencia de inmunorreactividad para la proteína S-100.
Caso 2
Niña de 9 años, con tumoración cutánea asintomática en la rodilla izquierda, de 1 mes de evolución. El diagnóstico clínico es de granuloma piogénico. Se recibe en el servicio de Anatomía Patológica una cuña cutánea de 0,8 x 0,6 cm con una lesión ligeramente elevada, abombada, de coloración pardusca, de aproximadamente 0,5 cm de diámetro. Se secciona por el eje mayor y se incluye en su totalidad para su estudio histológico.
Microscópicamente se identifica una tumoración dérmica mal delimitada, constituida por nidos y fascículos rodeados por un estroma colágeno denso (fig. 8). Las células tumorales muestran morfología epitelioide, un citoplasma ligeramente eosinófilo, y un núcleo vesiculoso, con pequeño nucléolo (fig. 9). Se observa frecuente englobamiento de pequeños nervios por las células tumorales, así como de vasos y glándulas sudoríparas (fig. 10). Hay una ligera cantidad de mucina azul-alcián positiva. El tumor respeta la dermis papilar y el tejido celular subcutáneo. La epidermis muestra ligera hiperqueratosis, acantosis e hiperpigmentación basal. En la dermis papilar se observa un ligero infiltrado inflamatorio linfocitario perivascular. No se ve atipia citológica ni fenómenos de necrosis. La tasa de mitosis es de 3 por 10 campos de gran aumento.
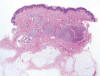
Fig. 8:
Tumoración dérmica constituida por nidos y fascículos rodeados por un estroma
colágeno denso.
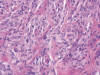
Fig. 9:
Células tumorales epitelioides, con citoplasma eosinófilo y núcleo vesiculoso
con pequeño nucléolo. Figura de mitosis en la parte superior izquierda.
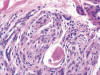
Fig. 10:
Englobamiento de un pequeño nervio por las células tumorales.
Las técnicas de inmunohistoquímica muestran positividad moderada-intensa para vimentina, CD68, CD10 y actina músculo-liso, y positividad discreta para factor XIIIa y enolasa neuronal específica. Son negativos el resto de marcadores, incluida la proteína S-100.
DISCUSIÓN
El neurotecoma celular es una tumoración cutánea benigna, poco frecuente, de histogénesis incierta. Afecta fundamentalmente a niños y adultos jóvenes, con un pico de incidencia en la segunda década. Es más frecuente en mujeres, en un ratio aproximado de 2:1 con respecto a los varones. Ocurre fundamentalmente en la parte superior del cuerpo, especialmente en la cara y el hombro (6,8). Ocasionalmente se describe en las extremidades inferiores, tórax, espalda, mucosa oral y conjuntiva.
La apariencia clínica es la de una tumoración solitaria, circunscrita, de crecimiento lento, asintomática y generalmente de pequeño tamaño (la mayoría son menores de 2 cm de diámetro) (6,8). Ocasionalmente se describen lesiones múltiples (10,20). El diagnóstico clínico es inespecífico. El tiempo de evolución oscila entre unas pocas semanas y varios años (6,8).
Microscópicamente, el neurotecoma celular se caracteriza por un patrón de crecimiento multinodular y una localización básicamente dérmica, aunque es muy frecuente la afectación del tejido celular subcutáneo superficial (6). Está constituido por nidos redondeados y ovalados de distinto tamaño, con frecuencia de apariencia arremolinada, rodeados por bandas de colágeno denso. A veces se observa una disposición focal de las células en fascículos. Generalmente hay una zona de dermis no afectada («grenz zone») entre la epidermis y el tumor. Las células tumorales muestran morfología fusiforme y epitelioide, citoplasma ligeramente eosinófilo, límites celulares imprecisos, y núcleo vesiculoso con nucléolo. Puede haber células multinucleadas de tipo osteoclástico, de Touton o tumorales. No son raras las figuras de mitosis. El índice mitótico medio es de 3 mitosis por 10 campos de gran aumento, aunque puede haber hasta más de 10 por 10 campos de gran aumento. Ocasionalmente se describen mitosis atípicas (6,8). La cantidad de matriz mixoide es generalmente escasa o totalmente ausente, y a menudo varía en cantidad de lóbulo a lóbulo. Sin embargo, se han descrito lesiones con abundante matriz mixoide e inmunofenotipo de neurotecoma celular (11). Estos tumores «mixtos» representan probablemente una variante mixoide de neurotecoma celular (8). Con frecuencia se observa un infiltrado inflamatorio linfocitario en el estroma, y a veces, entre las células tumorales. Más raramente puede haber células plasmáticas, eosinófilos o neutrófilos. Son comunes las células dendríticas presentadoras de antígenos, más evidentes mediante inmunohistoquímica (6).
Algunos neurotecomas celulares muestran células atípicas, a veces con marcado pleomorfismo nuclear, y numerosas figuras de mitosis. También se han descrito tumores de tamaño grande (hasta 6 cm de diámetro), con penetración profunda (extendiéndose hasta el músculo esquelético o tejido celular subcutáneo), con bordes difusamente infiltrativos, o con invasión vascular o perineural. La presencia de estas características histológicas «atípicas» no parece tener significado clínico (6,8,12). Sin embargo, es necesario el estudio de más casos con seguimiento a largo plazo para descartar la posibilidad de un comportamiento como un tumor maligno de bajo grado (12).
El neurotecoma celular tiene una tasa de recidiva baja. El mayor porcentaje de recidiva ocurre en lesiones en la cara de mujeres jóvenes, en las que generalmente el tratamiento inicial es más conservador y la biopsia no incluye tejido celular subcutáneo (6,8).
Desde el punto de vista inmunohistoquímico los neurotecomas celulares son típicamente positivos con NKI/C3, proteína S100A6, Mitf (factor transcriptor de microftalmia), PGP9.5, CD10 y vimentina, y variablemente positivos para enolasa neuronal específica, actina músculo-liso, factor XIIIa, CD68, sinaptofisina, cromogranina A, CD99 y PG-M1. Son negativos para la proteína S-100, proteína glial fibrilar acídica, HMB-45, melan A, EMA, citoqueratinas, CD34 y desmina (6,8,13-16).
Los hallazgos ultraestructurales indican que el neurotecoma celular está compuesto predominantemente por células incompletamente diferenciadas con características sugestivas de diferenciación divergente fibroblástica, miofibroblástica, muscular lisa y de células de Schwann (17).
Algunos autores sostienen que el neurotecoma celular muestra diferenciación neuroectodérmica, en base a la positividad inmunohistoquímica para marcadores neurales como PGP9.5 (16), NKI/C3 y Mitf (1), si bien se ha visto que estos marcadores tienen muy baja especificidad (11,18-21). También se ha descrito un caso con positividad para enolasa neuronal específica, sinaptofisina y cromogranina A, sugiriendo diferenciación neuroendocrina (22).
Se ha propuesto que el neurotecoma celular podría representar una variante epitelioide de leiomioma (23), una variante de dermatofibroma con peculiaridades arquitecturales y celulares (24), o un tipo de tumor mio-melanocítico de la familia de los tumores de células epitelioides perivasculares (pecomas) (15).
Otros autores encuadran al neurotecoma celular dentro del grupo de tumores «fibrohistiocitarios» (9). Misago y cols. describen diferenciación histiocítica en un neurotecoma celular, lo que según ellos podría indicar que es realmente un tumor fibrohistiocitario, o bien que es un tumor con diferenciación de vaina nerviosa inmadura con expresión de marcadores histiocíticos, o un tumor indiferenciado derivado de células de la cresta neural de linaje vaina nerviosa/fibrohistiocitario (25).
Hornick y Fletcher sugieren una diferenciación miofibroblástica (8), mientras que Fetsch y cols. proponen que el tumor deriva de células fibroblásticas con la capacidad de producir matriz mixoide y de diferenciarse en miofibroblastos, y con una tendencia de reclutar células histiocíticas (7).
Por último, algunos investigadores afirman que el neurotecoma celular podría estar relacionado con el tumor fibrohistiocítico plexiforme (9,26,27).
En los dos casos que presentamos, la positividad inmunohistoquímica para factor XIIIa, actina músculo-liso y CD68, sugiere diferenciación fibrohistiocitaria. La positividad para CD10 ya se ha descrito en dos casos de neurotecoma celular, que también eran positivos para CD68 (25,28), así como en todos los casos estudiados por Fetsch y cols. en su serie de 178 neurotecomas (7). Su significado no está claro. Se ha descrito positividad para CD10 en una gran variedad de neoplasias benignas y malignas, incluidas lesiones fibrohistiocitarias cutáneas (25,28).
El diagnóstico diferencial del neurotecoma celular hay que establecerlo fundamentalmente con el neurotecoma mixoide y con lesiones melanocíticas de patrón plexiforme (28).
Aunque el neurotecoma mixoide y el celular comparten algunas similitudes (fundamentalmente un crecimiento multilobulado dérmico y una mayor o menor cantidad de mucina estromal), el primero se caracteriza por la presencia de células fusiformes delgadas y estrelladas, más pequeñas, con núcleo citológicamente blando, interconectadas de forma laxa dentro de un abundante estroma mixoide. Las células del neurotecoma celular son más grandes, con frecuencia epitelioides, tienen un núcleo vesiculoso que puede exhibir algún grado de atipia, y se disponen en agregados arremolinados densos y a veces en fascículos, asociados en general a una escasa matriz mixoide, o con ausencia total de la misma. Inmunohistoquímicamente, el neurotecoma celular es típicamente S-100-negativo y NKI/C3-positivo, mientras que el neurotecoma mixoide muestra un inmunofenotipo opuesto (S-100-positivo y NKI/C3-negativo). Por otra parte, los neurotecomas mixoides tienen un pico de incidencia en la cuarta década de la vida, afectan casi en la misma proporción a varones y a mujeres, y tiene una marcada predilección por las extremidades, especialmente las manos (7). Los neurotecomas celulares, en cambio, tienen un pico de incidencia en la segunda década de la vida, un ratio varón-mujer de aproximadamente 1:2, y una distribución en la parte superior del cuerpo, sobre todo en la cara y el hombro. La distinción es clínicamente importante, ya que los mixomas de la vaina nerviosa tienen una tasa de recidiva local alta en caso de extirpación incompleta (7).
Las lesiones melanocíticas que pueden mostrar un patrón plexiforme incluyen el melanoma, el nevus de Spitz, el nevus de células fusiformes, el nevus penetrante profundo, el nevus azul celular, y más raramente el nevus congénito (28). La ausencia de componente juntural y la negatividad inmunohistoquímica para S-100 y HMB-45 en el neurotecoma celular ayudan a realizar el diagnóstico diferencial.
El tumor fibrohistiocítico plexiforme muestra marcada similaridad con el neurotecoma celular. Ambos están constituidos por células fusiformes y epitelioides, con un patrón de crecimiento multinodular, y pueden tener células gigantes de tipo osteoclasto. Sin embargo, el tumor fibrohistiocítico plexiforme tiene una localización predominantemente subcutánea, y muestra además fascículos de células fusiformes de tipo fibromatoso. La distinción es clínicamente relevante, ya que el tumor fibrohistiocítico plexiforme es una tumoración con comportamiento maligno de bajo grado.
Otras lesiones a tener en cuenta en el diagnóstico diferencial del neurotecoma celular son tumores musculares lisos, el dermatofibroma epitelioide, el reticulohistiocitoma, el tumor maligno de la vaina nerviosa periférica, el meningioma cutáneo, el perineuroma, el neurofibroma plexiforme, el schwannoma plexiforme y el tumor de células granulares plexiforme.
El neurotecoma celular no parece mostrar diferenciación neuroectodérmica, por lo que se justifica su separación del mixoma de la vaina nerviosa, y su posible inclusión dentro de los tumores «fibrohistiocíticos».
BIBLIOGRAFÍA
Harkin JC, Reed RJ, editores. Tumors of the peripheral nervous system. Atlas of tumor pathology. Second Series. Fascicle 3. Washington D.C.: Armed Forces Institute of Pathology; 1969. p. 60-4.
Gallager RL, Helwig EB. Neurothekeoma – a benign cutaneous tumor of neural origin. Am J Clin Pathol 1980; 74: 759-64.
Rosati LA, Fratamico FC, Eusebi V. Cellular neurothekeoma. Appl Pathol 1986; 4: 186-91.
Husain S, Silvers DN, Halperin AJ, McNutt NS. Histologic spectrum of neurothekeoma and the value of immunoperoxidase staining for S-100 protein in distinguishing it from melanoma. Am J Dermatopathol 1994; 16: 496-503.
Barnhill RL, Dickersin GR, Nickeleit V, Bhan AK, Muhlbauer JE, Phillips ME, Mihm MC Jr. Studies on the cellular origin of neurothekeoma: Clinical, light microscopic, immunohistochemical, and ultrastructural observations. J Am Acad Dermatol 1991; 25: 80-8.
Fetsch JF, Laskin WB, Hallman JR, Lupton GP, Miettinen M. Neurothekeoma: An analysis of 178 tumors with detailed immunohistochemical data and long-term patient follow-up information. Am J Surg Pathol 2007; 31: 1103-14.
Fetsch JF, Laskin WB, Miettinen M. Nerve sheath myxoma. A clinicopathologic and immunohistochemical analysis of 57 morphologically distinctive, S-100 protein- and GFAP-positive, myxoid peripheral nerve sheath tumors with a predilection for the extremities and a high local recurrence rate. Am J Surg Pathol 2005; 29: 1615-24.
Hornick JL, Fletcher CD. Cellular neurothekeoma: detailed characterization in a series of 133 cases. Am J Surg Pathol 2007; 31: 329-40.
Laskin WB, Fetsch JF, Miettinen M. The «neurothekeoma»: immunohistochemical analysis distinguishes the true nerve sheath mixoma from its mimics. Hum Pathol 2000; 31: 1230-41.
Scheithauer BW, Woodruff JM, Erlandson RA, editores. Tumors of the peripheral nervous system. Atlas of tumor pathology. Third Series. Fascicle 24. Washington D.C.: Armed Forces Institute of Pathology; 1999. p. 219-82.
Rudolph P, Schubert C. Myxoid cellular neurothekeoma. Am J Dermatopathol 2002; 24: 92-3.
Busam KJ, Mentzel T, Colpaert C, Barnhill RL, Fletcher CDM. Atypical or worrisome features in cellular neurothekeoma: a study of 10 cases. Am J Surg Pathol 1998; 22: 1067-72.
Fullen DR, Lowe L, Su LD. Antibody to S100a6 protein is a sensitive immunohistochemical marker for neurothekeoma. J Cutan Pathol 2003; 30: 118-22.
Mahalingam M, Alter JN, Bhawan J. Multiple cellular neurothekeomas – a case report and review on the role of immunohistochemistry as a histologic adjunt. J Cutan Pathol 2006; 33: 51-6.
Page RN, King R, Mihm MC Jr, Googe PB . Microphthalmia transcription factor and NKI/C3 expression in cellular neurothekeoma. Mod Pathol 2004; 17: 230-4.
Zelger BG, Steiner H, Kutzner H, Maier H, Zelger B. Cellular ‘neurothekeoma’: an epithelioid variant of dermatofibroma? Histopathology 1998; 32: 414-22.
Argenyi ZB, Kutzner H, Seaba MM. Ultrastructural spectrum of cutaneous nerve sheath mixoma/cellular neurothekeoma. J Cutan Pathol 1995; 22: 137-45.
Busam KJ, Iversen K, Coplan KC, Jungbluth AA. Analysis of microphthalmia transcription factor expression in normal tissues and tumors, and comparison of its expression with S-100 protein, gp100, and tyrosinase in desmoplastic malignant melanoma. Am J Surg Pathol 2001; 25: 197-204.
Campbell LK, Thomas JR, Lamps LW, Smoller BR, Folpe AL . Protein gene product 9.5 (PGP 9.5) is not a specific marker of neural andl nerve sheath tumors: An immunohistochemical study of 95 mesenchymal neoplasms. Mod Pathol 2003; 16: 963-9.
Granter SR, Weilbaecher KN, Quigley C, Fletcher CDM, Fisher DE. Microphthalmia transcription factor. Not a sensitive or specific marker for the diagnosis of desmoplastic melanoma and spindle cell (non-desmoplastic) melanoma. Am J Dermatopathol 2001; 23: 185-9.
Sachdev R, Sundram UN. Frequent positive staining with NKI/C3 in normal and neoplastic tissues limits its usefulness in the diagnosis of cellular neurothekeoma. Am J Clin Pathol 2006; 126: 554-63.
Chang SE, Lee TJ, Ro JY, Choi JH, Sung KJ, Moon KC, Koh JK. Cellular neurothekeoma with possible neuroendocrine differentiation. J Dermatol 1999; 26: 363-7.
Calonje E, Wilson-Jones E, Smith NP, Fletcher CD. Cellular ‘neurothekeoma’: an epitelioid variant of pilar leiomyoma? Morphological and immunohistochemical analysis of a series. Histopathology 1992; 20: 397-404.
Zelger BG, Steiner H, Kutzner H, Maier H, Zelger B. Cellular ‘neurothekeoma’: an epithelioid variant of dermatofibroma? Histopathology 1998; 32: 414-22.
Misago N, Satoh T, Narisawa Y. Cellular neurothekeoma with histiocytic differentiation. J Cutan Pathol 2004; 31: 568-72.
Jaffer S, Eusebi V, Rosai J. Neurothekeomas and plexiform fibrohistiocytic tumors: a relationship?. Lab Invest 2000; 80: 11A.
Requena L, Sangüeza OP. Benign neoplasms with neural differentiation: A review. Am J Dermatopathol 1995; 17: 75-96.
Alkhalidi H, Ghazarian D. Cellular neurothekeoma with a plexiform morphology: a case report with a discussion of the plexiform lesions of the skin. J Cutan Pathol 2007; 34: 264-9.
![]()