
Vol. 41, n.º 3, 2008
|
REVISTA
ESPAÑOLA DE
Vol. 41, n.º 3, 2008 |
CASUÍSTICA
Manuel Vaquero
Hospital Donostia de San Sebastián.
RESUMEN
Introducción: El sarcoma fibroblástico mixoinflamatorio es una lesión mesenquimal rara pero característica que se localiza en el tejido celular subcutáneo y que puede confundirse con procesos inflamatorios o infecciosos, linfoma de Hodgkin y diversos sarcomas. Caso clínico: Se describe un caso en un hombre de 62 años, localizado en la rodilla y tratado con escisión amplia, que precozmente recurrió y dio metástasis en pulmón y ganglios linfáticos regionales sin obtenerse respuesta ni con radioterapia local ni quimioterapia. Macroscópicamente el tumor tenía aspecto multinodular y bordes infiltrantes. Histológicamente se reconocía un patrón vagamente nodular con zonas inflamatorias y transición a otras mixoides o fibroesclerosantes. Se reconocieron células tumorales típicas de hábito hodgkiniano o virocítico. Las técnicas de inmunohistoquímica no fueron útiles para el diagnóstico. El paciente está vivo con enfermedad dos años después.
Palabras clave: Sarcoma fibroblástico mixoinflamatorio, tumores mixoides, sarcomas de partes blandas.
SUMMARY
Introduction: The myxoinflammatory fibroblastic sarcoma is a rare but distinctive mesenchymal lesion. This tumor develops in subcutaneous tissue and can be mistaken for an inflammatory or infectious process, Hodgkin’s lymphoma, or various sarcomas. Case report: A case in a 62 year-old man is described with early tumor recurrence and lung and regional lymph-node metastases after wide local excision without response to local radiotherapy and systemic chemotherapy. Grossly, the tumor was nodular with infiltrative margins. Histologically, the architecture was vaguely nodular with dense inflammatory infiltrate merging with mixoid or fibrosclerotic areas. Typical tumoral Hodgkin-like or «virocyte» cells were found. Inmunohistochemical stains were not useful in diagnosis. The patient is alive with disease two years later.
Keywords: Myxoinflammatory fibroblastic sarcoma, myxoid tumors, soft tissue sarcomas.
INTRODUCCIÓN
El sarcoma fibroblástico mixoinflamatorio (SFMI) fue descrito con diferentes denominaciones en 1998 por tres grupos diferentes de patólogos (1-3). En todas ellas se resaltaban su localización preferente en la parte distal de las extremidades y su tendencia a las recidivas. Histológicamente destacaban su multinodularidad y la mezcla de componentes tumorales mixoide, hialino-fibroso y epitelioide-fusocelular, junto a zonas inflamatorias y la presencia de células con nucleolos prominentes de aspecto hodgkiniano o virocítico.
Desde entonces han habido muy pocas comunicaciones en la literatura. En una se resalta la localización del tumor en el cuello (4) y en otras las alteraciones citogenéticas (5,6) o los hallazgos observados en la punción aspiración con aguja fina (PAAF) (7). El objetivo del trabajo es comentar un paciente con un SFMI localizado en una extremidad y que, a diferencia de lo habitual, cursó precozmente con metástasis ganglionares demostradas histológicamente y probablemente pulmonares.
DESCRIPCIÓN DEL CASO
Un varón de 62 años consulta por una tumoración palpable en la zona interna de la rodilla izquierda que había notado desde hacía varios meses. El resto de la exploración clínica fue normal. La TAC de la zona demostró una tumoración de 15 cm, con nódulos de varias densidades, que se extendía desde la dermis hasta los tejidos profundos, contactando con el plano vasculonervioso de la pierna (fig. 1). No había daño óseo ni ulceración. La TAC sistémica no demostró otras lesiones. Una primera biopsia incisional superficial no fue suficiente para asegurar un diagnóstico. Una muestra más profunda mostró los rasgos característicos del SFMI. Se extirpó el tumor con los márgenes más amplios posibles dada su localización en la rodilla y la proximidad del paquete vasculonervioso. En el extremo superior de la pieza habían 4 cm de músculo sin afectación pero en el plano profundo el tumor alcanzaba el plano fascial, aunque no se observó infiltración microscópica de los bordes quirúrgicos.
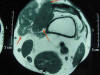
Fig. 1:
TAC de la rodilla en el que se observa una masa heterogénea que alcanza los
planos profundos.
El tumor recurrió localmente a los cuatro meses y causó un marcado agrandamiento ganglionar regional dos meses más tarde, que la biopsia demostró se correspondían con metástasis.
Morfológicamente la tumoración alcanzaba la dermis reticular y el patrón de crecimiento era vagamente nodular (fig. 2). Se visualizarón tres componentes: mixoide, fibrohialino e inflamatorio. El mixoide ocupaba la zona de los nódulos y contaba con células gigantes tumorales multinucleadas peculiares (fig. 3); el fibrohialino tenía moderada celularidad epitelioide-fusiforme (fig. 4), con 2 mitosis/10 CGA y mínima atipia, excepto en dos pequeñas zonas con atipias muy evidentes y mayor número de mitosis. El componente inflamatorio era el menos llamativo y tenía una distribución irregular. Predominaban los linfocitos maduros con algunos eosinófilos y escasas células grandes con núcleos uni o poli lobulados y nucleolos llamativos (fig. 5). No había necrosis. La inmunohistoquímica únicamente resultó positiva para vimentina y, focalmente, para CD34 (figs. 3 y 4, recuadros). Los marcadores habituales de músculo liso (SMA), célula de Schwann (S-100), células de Hodgkin (CD30, CD15) y ALK y pancitoqueratinas AE1-AE3 fueron negativos.
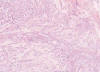
Fig. 2:
Nodularidad de la tumoración y aspecto mixoide.

Fig. 3:
Aspecto peculiar de las células gigantes tumorales de las áreas mixoides.
Recuadro: CD34 en esas células.
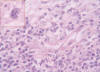
Fig. 4:
Área inflamatoria con ocasionales células estembergoides (recuadro).
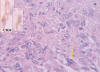
Fig. 5:
Celularidad epitelioide pleomórfica (flecha) y positividad para CD 34
(recuadro).
A los cuatro meses de la escisión quirúrgica con márgenes libres, el paciente estaba sin enfermedad pero dos meses más tarde mostró una recidiva local y un agrandamiento marcado de los ganglios inguinales ipsilaterales. La biopsia de la zona inguinal demostró un tumor con las mismas características que el de la rodilla. En el TAC realizado posteriormente se constaron nódulos pulmonares sospechosos de metástasis, por lo que se añadió quimiterapia sistémica sin obtenerse ninguna respuesta. Dos años después, a pesar de un nuevo intento de escisión que resultó con márgenes afectados y de radioterapia local, las masas inguinales se han extendido por el eje iliofemoral ipsilateral y han empeorado las imágenes pulmonares.
DISCUSIÓN
El SFMI (8-13) afecta por igual a ambos sexos y puede aparecer desde la primera década de la vida, aunque predomina en torno a la cuarta década. Es un tumor raro, con 128 casos descritos en la actualidad, de los que casi la centena se describieron en las tres comunicaciones iniciales. El tumor presenta variaciones morfológicas importantes según la proporción de los distintos componentes. A nivel ultraestructural las células tumorales recuerdan a fibroblastos (13). Hay casos con predominio de linfocitos y eosinófilos que obligan al diagnóstico diferencial con una enfermedad de Hodgkin, con infecciones virales o con la enfermedad de Rosai-Dorfman, aunque pueden ser predominantes las áreas mixoides y confundirse con liposarcomas mixoides, sarcomas fibromixoide de bajo grado o con mixofibrosarcomas. La localización acral y las áreas epitelioides recordarían a un sarcoma epitelioide pero no hay necrosis y las queratinas son negativas. En el presente caso la primera biopsia incluía la zona de hipodermis infiltrada por el tumor por lo que la cantidad de adipocitos y el aspecto mixoide recordaban a un liposarcoma, aunque no cumplía todos los criterios diagnósticos, por lo que se recomendó una biopsia de zonas más profundas que permitió realizar el diagnóstico definitivo. Hay publicado un estudio con PAAF (7) de tres casos en los que sólo uno no tenía los diversos componentes del tumor. Sin embargo, la gran variabilidad de unas zonas a otras y la escasez de alguno de sus componentes en bastantes casos publicados hacen pensar que esta técnica no es la idónea para orientar el diagnóstico. La inmunohistoquímica tampoco es de gran ayuda dada la negatividad o mínima positividad focal de los anticuerpos que frecuentemente se utilizan en las lesiones de partes blandas. También está descrita la positividad con CD68 y, débilmente, de citoqueratinas (13). La b-catenina también resultó negativa en un único caso analizado (14). En el presente tumor la positividad con CD34 fue focal pero intensa y en todo tipo de células neoplásicas. En cierto sentido semejante a la observable excepcionalmente en sarcomas fibromixoides de bajo grado (13) y en algunas proliferaciones fibroblásticas-miofibroblásticas que nunca alcanzan la intensa positividad del dermatofibrosarcoma, el tumor fibroso solitario o el miofibrosarcoma (15). En cuanto a las células tumorales llamadas virocíticas recuerdan a los fibroblastos presentes infrecuentemente en el corion de vejigas urinarias con irritación crónica o en el conectivo en torno a lesiones inflamatorias crónicas, como el que rodea las prótesis óseas.
Hay dos SFMI con estudios citogenéticos. Lambert et al (5) encuentran un cariotipo complejo con una translocación t(1;10) (p22;q24) y pérdida de los cromosomas 3 y 13. Por su parte Mansoor et al (6) describen, por técnica de FISH, cromosomas en anillo de diverso tamaño y complejidad y con material del cromosoma 3, considerándolo como una forma peculiar de amplificación del material genético. Los cromosomas en anillo de estructura compleja, con marcada variación en su tamaño y número, se han descrito asociados a tumores mesenquimales de bajo grado de malignidad como el dermatofibrosarcoma protuberans, el osteosarcoma paraostal y el tumor lipomatoso atípico (16).
Las recidivas locales ocurren en más de la mitad de los pacientes y en la actualidad no hay criterios morfológicos o inmunohistoquímicos, incluidos la p53 y el MIB-1 (10), que permitan discernir entre los casos que sufrirán recaídas y los que no. Tan sólo hay publicados cuatro casos con metástasis, tan sólo uno con localización extranodal (2). Se recomienda una excisión completa con los márgenes más amplios posibles. En el presente caso, el gran tamaño tumoral y su localización en la rodilla que dificultó la cirugía conservadora con márgenes amplios podrían explicar la mala evolución posterior de la neoplasia.
BIBLIOGRAFÍA
Montgomery EA, Davaney KO, Giordano TJ. Inflammatory mixohyaline tumor of distal extremities with virocyte or Reed-Sternbarg-like cells: A distinctive lesion with features simulating inflammatory conditions, Hodgkins disease, and varius sarcomas. Mod Pathol 1998; 11: 384-91.
Meis-Kindblom JM, Kindblom LG, . Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol 1998; 22: 911.
Michal M. Inflammatory myxoid tumor of the soft parts with bizarre giant cells. Pathol Res Pract 1998; 194: 529.
McFarlane R, Meyers AD, Glolitz L. Mixoinflammatory fibroblastic sarcoma of the neck. J Cutan Pathol 2005; 32: 375-8.
Lambert I, Debrec-Rychter M, Guenlinckx P, Hamemeijei A, Sciot R. Acral myxoinflammatory fibroblastic sarcoma with unique clonal chromosomal changes. Virchows Arch 2001; 438: 509.
Mansoor A, Fidda N Himoe E, Payne M, Lawce H, Magenis E. Myxoinflammatory fibroblastic sarcoma with complex supernumerary ring chromosomes composed of chromosome 3 segments. Cancer Genet Cytogenet 2004; 152: 61-5.
Pohar-Marinsek Z, Flezar M, Lamovec J. Acral muxoinflammatory fibroblastic sarcoma in FNAB samples: can we distinguish it from other myxoid lesions? Cytopathology 2003; 14: 73-8.
Kindblom LG, Meis-Kindblom JM. Myxoinflammatory fibroblastic sarcoma. En: DM Fletcher, KK Uni, F Mertens, editores. World Health Organization classification of tumors: Pathology and genetics of tumours of soft tissue and bone. Lyon: IARC Press; 2002. p. 96.
Jursic V, Zidar A, Montiel MD et al. Myxoinflammatory fibroblastic sarcoma: a tumor not restricted to acral sites. Ann Diag Pathol 2002; 6: 272.
Sakaki M, Hirokawa M, Watatsuki S, Sano T, Endo K et al. Acral myxoinflammatory fibroblastic sarcoma: a report of five cases and review of the literature. Virchow Arch 2003; 442: 25-30.
Genevay M, Coindre JM, Guillon L. Entités récentes en pathologie tumorale des tissue mous (2e partie). Ann Pathol 2003; 23: 135-48.
Lang JE, Dodd L, Martinez S, Brigman BE. Acral myxoinflammatory fibroblastic sarcoma. A report of five cases and literature review. Clin Orthop Relat Res 2006; 445: 254-60.
Kindblom LG, Meis-Kidblom JM. Myxoinflamatory fibroblastic sarcoma. En: Fletcher CDM, Unni KK, Mertens F., editors. Pathology and Genetic of tumors of soft tissue and bone (WHO). Lyon: IARC Press; 2002. p. 96-7.
Bhattacharya B, Dilwoth HP, Iacobuzco-Donahue C, Ricci F, Weber K, Turlog MA, Fisher C, Montgomery E. Nuclear ‚-catenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic lesions. Am J Surg Pathol 2005; 29: 653-9.
Fisher C. Low-grade sarcomas with CD34-positive fibroblasts and low-grade myofibroblastic sarcomas. Ultrastructural Pathol 2004; 28: 291-305.
Pendentour F, Foms A, coindre JM, Berne JM, Nicolo G, Mucjiels JF, et al. Structure of the supernumerary ring and grand rod chromosomes in adipose tumors. Genes Chromosomes Cancer 199; 24: 30-41.
![]()