
Vol. 41, n.º 3, 2008
|
REVISTA
ESPAÑOLA DE
Vol. 41, n.º 3, 2008 |
CASUÍSTICA
Francisco Javier Torres Gómez, Rafael García Ligero Ochoa, Fernando Martínez de Salazar Bascuñana
Servicio de Anatomía Patológica. Hospital Punta
de Europa. Carretera Getares, s/n, 11207 Algeciras.
javiertorresgomez@yahoo.es
RESUMEN
Antecedentes: El paracordoma es una lesión inocua de tejidos blandos que muestra unas características histológicas similares a las del cordoma axial. Método: Presentamos un caso de paracordoma haciendo hincapié en sus rasgos histológicos y en su diagnóstico diferencial. Resultados: Se trata de una lesión sin capacidad para metastatizar cuyo tratamiento es la simple extirpación quirúrgica. Conclusiones: Hay que pensar en esta entidad ante un tumor mixto de tejidos blandos superficiales.
Palabras clave: Paracordoma, cordoma extraaxial, células fisalíferas.
SUMMARY
Introduction: parachordoma is a benign lesion of soft tissues that shows histological features resembling those of axial chordoma. Material and methods: We present a case of parachordoma, highlighting histological features and differential diagnosis. Results: Parachordoma is a non-metastasizing lesion successfully treated by simple surgical removal. Conclusions: Parachordoma is included in differential diagnosis of mixed superficial soft tissue tumours.
Keywords: Parachordoma, extraaxial chordoma, physaliferous cells.
INTRODUCCIÓN
A pesar de ser una lesión infrecuente, los paracordomas son lesiones características desde el punto de vista histológico pues recuerdan a los cordomas axiales. De hecho, son lesiones superponibles desde el punto de vista morfológico, residiendo su diagnóstico diferencial principalmente en la localización. Se trata de lesiones benignas sin capacidad metastatizante cuyo tratamiento consistirá exclusivamente en la exéresis quirúrgica.
CASO CLÍNICO
Paciente varón de 46 años que presentaba una lesión nodular en región costal izquierda de 2 cm de dimensiones máximas, bien delimitada y de consistencia elástica-firme que según nos fue referido había crecido progresivamente en los últimos meses sin causar sintomatología. Se extirpó la lesión la cual, a los cortes seriados, mostraba una tonalidad predominantemente amarillenta a modo de nódulos a nivel central y un aspecto blanquecino y fibroso en periferia. Existían así mismo focos con aspecto de calcificación distribuidos al azar en la lesión. El estudio microscópico permitió observar una buena delimitación lesional estando rodeada por tejido conectivo fibroso denso. El núcleo lesional estaba constituido por células poligonales de un tamaño mediano-grande con un amplio citoplasma claro y un núcleo vesiculoso, ovoideo, reniforme o polilobulado dotado de un nucleolo de tamaño mediano. Dichas células se disponían formando ductos, cordones e incluso hileras separadas por un denso estroma fibroso que en ocasiones adoptaba un aspecto hialino. En otras áreas las células mostraban una morfología fusocelular. La actividad mitósica observada era muy escasa (figs. 1 a 5). El estudio inmunohistoquímico de la población celular lesional demostró positividad para pancitoqueratinas, S100, EMA, vimentina y positividad débil y focal para actina; la desmina en cambio resultó negativa. Con estos hallazgos y con la característica histología de la lesión pudimos emitir el diagnóstico de paracordoma (mioepitelioma de tejidos blandos de Fletcher).
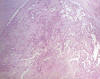
Fig. 1:
Paracordoma. Imagen panorámica. HE-100x.
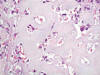
Fig. 2:
Paracordoma. Detalle de la matriz condromixoide. HE-400x.
 -
-
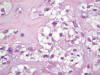
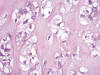
Figs. 3 y 4:
Paracordoma. Células fisalíferas. Detalle. HE-400x.
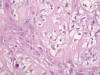
Fig. 5:
Paracordoma. Detalle histológico. HE-200x.
Los paracordomas son tumores muy poco frecuentes, descritos por primera vez por Dabska (1) cuya histogénesis es controvertida. Afectan a un amplio rango de edades y a ambos sexos si bien los varones se afectan algo más que las mujeres. La localización más frecuente es a nivel de los tejidos blandos de las extremidades, originando una masa nodular de tamaño variable y crecimiento lento que no suele preocupar al paciente, el cual, suele consultar tras un determinado lapso de evolución. Le siguen en frecuencia la localización en cabeza y cuello y a nivel torácico, como en nuestro caso (2,3).
Como hemos comentado se trata de una entidad no exenta de controversia; ésta se focaliza principalmente en la distinción o no del cordoma periférico o extraesquelético y del paracordoma (4,5). Si bien son muchos los autores que consideran estos términos sinónimos, son también muchos los que diferencian estas dos entidades basándose en criterios inmunohistoquímicos y de evolución (6,7). Así, el cordoma periférico sería una lesión exactamente igual que la descrita a nivel axial con la única diferencia de la localización; por tanto existiría la posibilidad no sólo de recidivas lesionales sino también de un comportamiento metastático. De este modo, aquellos escasos ejemplos en que se han documentado metástasis entrarían a formar parte de este grupo. Los paracordomas en cambio carecerían por definición de este potencial, siendo lesiones inocuas en todos los casos. También se han pretendido encontrar diferencias en cuanto a las propiedades de tinción inmunohistoquímica de ambas entidades de forma que sólo los cordomas se teñirían para queratinas de alto peso molecular y CEA mientras que las de bajo peso, EMA y S100 serían comunes a ambas. La controversia está servida en el momento que existen estudios que no logran reproducir estas diferencias. Con un nombre u otro se trata de una lesión con una característica histología demostrándose una población de células poligonales claras que recuerdan a las de la notocorda y una segunda población de células eosinofílicas, ambas organizadas en cordones y nidos de distintos tamaños observándose en ocasiones grupos celulares con morfología fusiforme o pequeñas. Asimismo es frecuente observar fenómenos de vacuolización citoplasmática que confieren a la lesión un gran parecido con el cordoma (células fisalíferas); al igual que en este último es infrecuente apreciar tanto pleomorfismo celular como figuras mitóticas y el estroma adquiere características mixoides o hialinas (8-10).
Si bien la lesión está aparentemente bien circunscrita, el estudio detallado de la misma permite observar nidos tumorales, generalmente de pequeño tamaño, infiltrando los tejidos adyacentes. Es precisamente esta característica la responsable de las recurrencias locales descritas en esta entidad no habiéndose descrito metástasis a distancia (los casos que describen metástasis han sido desestimados como auténticos paracordomas) (11). Obviando las recurrencias descritas se trata de neoplasias benignas para las que la exéresis quirúrgica es el tratamiento adecuado sin que sea necesario recurrir a tratamientos más agresivos o adyuvancia para su manejo. En nuestro caso el paciente no ha referido recidivas en los cuatro años siguientes a la extirpación de la lesión. El paracordoma es una entidad actualmente reconocida y prueba de ello es su inclusión en la última clasificación de la Organización Mundial de la Salud si bien se encuentra formando parte del grupo Tumor Mixto/ Mioepitelioma/ Paracordoma donde se engloban variantes de lo que parece ser una única entidad con distintos fenotipos. Desde este punto de vista el paracordoma sería una variante de tumor mixto de tejidos blandos y no nos debe de sorprender tal explicación pues el estroma condromixoide y la variada celularidad presente en este tipo de lesiones bien pueden recordar a la de similares lesiones de la glándula salival existiendo, eso sí, una serie de propiedades que le permiten adquirir personalidad propia. Las peculiares características morfológicas del paracordoma pueden en un momento dado presentar dudas diagnósticas existiendo varias entidades con las que puede plantearse un diagnóstico diferencial que en la mayoría de las ocasiones es más teórico que real pues la clínica ayuda a hacer la oportuna distinción. Una de estas entidades es el condrosarcoma de células claras. El estroma mixoide-condromixoide del paracordoma así como la presencia de células claras vacuolizadas puede ser una característica común a ambas lesiones si bien la edad y localización de las mismas así como una cuidadosa búsqueda de atipia celular ayudan a resolver el problema (12). Otra entidad a tener en cuenta es la metástasis de un carcinoma de células claras. La atipia celular y el examen sistémico del paciente para localizar el primario serán de gran importancia en este cometido. Son varios los trabajos que han intentado caracterizar al paracordoma desde el punto de vista genético, habiéndose detectado anomalías tanto estructurales como numéricas de tipo clonal, siendo las más frecuentes t(2,4), del(3q) o las pérdidas de cromosomas 9, 10, 20 y 22. Otros trabajos difieren de estos resultados y es que la infrecuencia de este tipo de lesiones quizá requiera de más tiempo o de la recogida de un número suficiente de casos para que los resultados puedan ser estandarizados (13,14).
BIBLIOGRAFÍA
Dabska M. Parachordoma: a new clinicopathologic entity. Cancer 1977; 40: 1586-92.
Gimferrer JM, Baldo X, Montero CA, Ramirez J. Chest wall parachordoma. Eur J Cardiothorac Surg 1999; 16: 573-5.
Tong G, Perle MA, Desai P, Kumar A, Waisman J. Parachordoma or chordoma periphericum? Case report of a tumor of the thoracic wall. Diagn Cytopathol 2003; 29: 18-23.
Scolyer RA, Bonar SF, Palmer AA, Barr EM, Wills EJ, Stalley P, et al. Parachordoma is not distinguishable from axial cordoma using immunohistochemistry. Pathol Int 2004; 54: 364-70.
Fisher C. Parachordoma exists but what is it? Adv Anat Pathol 2000; 7: 141-8.
van Akkooi AC, van Geel AN, Bessems JH, den Bakker MA. Extraaxial chordoma. J Bone Joint Surg Br 2006; 88: 1232-4.
Separoviç R, Glumbiç I, Pigac B, Separoviç V, Kruslin B. Parachordoma: a case report. Tumori 2001; 87: 207-10.
Sangueza OP, White CR Jr. Parachordoma. Am J Dermatopathol 1994; 16: 185-8.
Alfieri S, Prete FP, Di Giorgio A, Doglietto GB. Retroperitoneal parachordoma in a patient with a history of recurrent pain. Lancet Oncol 2005; 6: 350.
Nielsen GP, Mangham DC, Grimer RJ, Rosenberg AE. Chordoma periphericum: a case report. Am J Surg Pathol 2001; 25: 263-7.
Abe S, Imamura T, Harasawa A, Ishida T, Unno K, Tateishi A, et al. Parachordoma with multiple metastases. J Comput Assist Tomog 2003; 27: 634-8.
Folpe AL, Agoff SN, Willis J, Weiss SW. Parachordoma is immunohistochemically and cytogenetically distinct from axial chordoma and extraskeletal myxoid chondrosarcoma. Am J Surg Pathol 1999; 23: 1059-67.
Tihy F, Scott P, Russo P, Champagne M, Tabet JC, Lemieux N. Cytogenetic analysis of a parachordoma. Cancer Genet Cytogenet 1998; 105: 14-9.
Limon J, Babi´nska M, Denis A, Ry´s J, Niezabitowski A. Parachordoma: a rare sarcoma with clonal chromosomal changes. Cancer Genet Cytogenet 1998; 102: 78-80.
![]()