
Vol. 41, n.º 3, 2008
|
REVISTA
ESPAÑOLA DE
Vol. 41, n.º 3, 2008 |
REVISIONES
José I. López1, Aitziber Ugalde2, Ming Zhou3
Hospital de Cruces-Osakidetza, Plaza de Cruces,
s/n.
1 Departamento de
Anatomía Patológica. Hospital de Cruces. Universidad del País Vasco (EHU/UPV).
Plaza Cruces-gurutzeta, 12. 48902 Barakaldo, Bizkaia.
joseignacio.lopez@ehu.es
2 Servicio de
Anatomía Patológica. Hospital de Basurto. Bilbao. Bizkaia.
aitziber.ugaldeolano@osakidetza.net
3 Department of
Pathology, Microarray Unit. The Cleveland Clinic Foundation. Cleveland, OH,
EEUU. zhoum@ccf.org
RESUMEN
En este trabajo se revisa el concepto del carcinoma renal constituido por células claras, un hecho muy frecuentemente encontrado en la práctica diaria en nuestro medio. Este abordaje es pertinente debido a que no todos los carcinomas renales con células claras al microscopio son auténticos carcinomas renales de células claras, sino que pueden ser otras entidades. Dicha aproximación puramente morfológica no reniega de la nueva clasificación de la OMS vigente, sino que la complementa. El trabajo está dirigido más al patólogo general que se enfrenta a casos muy variados con alta presión asistencial, que al subespecialista acostumbrado a profundizar en casos complejos y con grandes medios técnicos en el campo del cáncer renal. Se revisa en primer lugar el carcinoma renal asociado a la enfermedad de von Hippel-Lindau porque muchas de las alteraciones genéticas que provocan esta enfermedad están presentes en el carcinoma renal de células claras esporádico y con posterioridad, se hace lo mismo con los carcinomas renales de células claras esporádicos convencionales, los carcinomas familiares no asociados a la enfermedad de von Hippel-Lindau, los carcinomas con translocación, el carcinoma papilar renal hereditario, algunos carcinomas asociados a Esclerosis Tuberosa y al síndrome de Birt-Hogg-Dubé, el carcinoma de células cromófobas y el carcinoma asociado a enfermedad renal quística.
Palabras clave: Carcinoma renal, células claras, alteración genética, diagnóstico, diagnóstico diferencial.
SUMMARY
We review in this paper the concept of renal cell carcinoma with clear cells, a quite frequent histological finding in the pathologist’s daily work. This viewpoint is pertinent because not all the renal cell carcinomas with clear cells are conventional clear cell renal cell carcinomas. In fact, some of them are other entities on the light of the new WHO classification. This purely morphological approach does not dissent from this classification of renal tumours in adults. The revision is addressed more to the general pathologist who usually faces a wide variety of cases often under clinical pressure than to the subspecialist, who in general analyzes many complex cases of renal cancer in depth using sophisticated technical tools. Renal cell carcinoma related to von Hippel-Lindau disease is reviewed first because many of the genetic disorders underlying this disease are also present in sporadic, conventional renal cell clear cell carcinoma. Latter, conventional renal cell clear cell carcinomas, other familial, non von Hippel-Lindau associated renal cell carcinomas, translocation carcinomas, the hereditary papillary renal cell carcinoma, some carcinomas associated to Tuberous Sclerosis and to Birt-Hogg-Dubé syndrome, chromophobe renal cell carcinomas and carcinomas associated to end-stage renal disease are reviewed.
Keywords: Renal cancer, clear cell, genetic disorder, diagnosis, differential diagnosis.
INTRODUCCIÓN
Los patólogos sabemos bien de lo que hablamos cuando nos referimos a las células claras porque convivimos con ellas durante toda nuestra vida profesional. «Célula clara», al igual que «célula pequeña», «célula redonda», y otras «células», son expresiones de uso tan común en la práctica diaria que pertenecen al acervo del patólogo desde su primera semana de residencia. El término hace referencia al característico citoplasma claro o vacío, no teñido con la hematoxilina-eosina, de algunas células. Por lo general el citoplasma suele además estar delimitado por una membrana muy evidente. Un amplio espectro de neoplasias, y no sólo carcinomas, pueden contener eventualmente células claras. Bastará echar un vistazo al índice alfabético de cualquier libro de patología quirúrgica al uso (1,2), o teclear «clear cell carcinoma» en PubMed, para darse cuenta de lo larga y variada que puede ser la lista.
En ocasiones, la célula clara confiere al tumor un carácter exclusivo y lo convierte en entidad propia con pronóstico incorporado y en otras, la apariencia clara de la célula es simplemente un cambio secundario, una mera circunstancia sin ningún tipo de implicaciones en el diagnóstico o en el pronóstico. Conviene, pues, distinguir el carcinoma con células claras y el carcinoma de células claras, porque ambos términos no son necesariamente sinónimos.
Dicho esto, hay que resaltar que muchos órganos pueden desarrollar carcinomas de células claras, aunque el ovario y el riñón son dos de los más frecuentes. En este trabajo se revisa el espectro de carcinomas renales que pueden mostrar células claras al microscopio, a saber, el carcinoma renal asociado a la enfermedad de von Hippel-Lindau, el carcinoma renal de células claras de tipo esporádico, los carcinomas familiares no asociados a la enfermedad de von Hippel-Lindau, los carcinomas con translocación, y algunos carcinomas asociados a Esclerosis Tuberosa.
CARCINOMA RENAL ASOCIADO A LA ENFERMEDAD DE VON HIPPEL-LINDAU
Historia
La enfermedad de von Hippel-Lindau (VHL) es un síndrome neoplásico hereditario caracterizado por hemangioblastomas retinianos y del sistema nervioso central, carcinoma renal de células claras, y feocromocitoma. Afecta aproximadamente a 1 de cada 35.000 personas de manera autosómica dominante. Fue descrito en 1894 por Treacher Collins al describir las lesiones vasculares en la retina de dos gemelos. Sin embargo tomó su nombre del oftalmólogo alemán Eugene von Hippel y del patólogo sueco Arvid Lindau que estudiaron, cada un por separado y una década más tarde que el doctor Collins, las lesiones retinianas y cerebelosas de la enfermedad.
A partir de ahí, se han descrito una serie de lesiones asociadas a esta enfermedad, en especial quistes viscerales (pancreáticos y renales), y una serie amplia de tumores (carcinomas renales de células claras, feocromocitomas adrenales, tumores del saco endolinfático, del ligamento ancho, y del epidídimo, y tumores de los islotes pancreáticos) (3).
Genética
El gen responsable de la VHL fue localizado y descrito por primera vez en 1988 por Seizinger y cols. (4) en el brazo corto del cromosoma 3. Consta de 3 exones y se expresa de manera ubicua, y no sólo en los órganos asiento de tumores o lesiones.
La inactivación en línea germinal de uno de los alelos del gen VHL es la causa del síndrome neoplásico, el cual sale a la luz cuando se produce la mutación somática del otro alelo. Las mutaciones del gen VHL son extraordinariamente heterogéneas y están distribuidas a lo largo de toda la secuencia del gen. Se han descrito hasta la fecha unas 150 mutaciones diferentes (5). La lista de las mismas se puede consultar en http://www.umd.necker.fr:2005 y en http://web.ncifcrf.gov
El gen VHL es un gen supresor. Codifica una proteína con dos isoformas, pVHL30 y pVHL19 responsables de la acción antitumoral del gen. En condiciones normales, la proteína produce la inactivación de los factores inductores de hipoxia (FIH). Su ausencia, por el contrario, hace que estos FIH se estabilicen en la célula e induzcan la activación transcripcional de una amplia serie de factores reguladores de la angiogénesis, del ciclo celular y del crecimiento celular, por ejemplo, el factor de crecimiento endotelial vascular y la eritropoyetina y el factor de crecimiento derivado de las plaquetas, entre otros. La presencia de estos factores en el medio son los que en definitiva llevan al desarrollo de los tumores y quistes típicos de esta enfermedad (6,7). El conocimiento de este mecanismo genético de producción de la enfermedad abre las puertas a posibles tratamientos a través de los modernos modelos de terapia génica.
Sin embargo, pVHL también tiene un papel en la regulación de las metaloproteinasas y de sus inhibidores, y de otras enzimas. Por ejemplo, la ausencia de pVHL incrementa los niveles de las anhidrasas carbónicas 9 y 12, sustancias que incrementan el nivel de acidificación del microambiente, lo cual favorece a su vez las propiedades invasoras y el crecimiento tumoral, y sobrerregulan la ciclina D1 (7). La presencia de anhidrasa carbónica 9 (CA9) y de ciclina D1 puede se ser determinada por métodos inmunohistoquímicos en cortes de parafina, lo cual es importante para el patólogo en la practica diaria como veremos después.
El carcinoma renal de células claras asociado a la enfermedad de von Hippel-Lindau suele ser con frecuencia multifocal, bilateral y quístico, y aparece en edades más tempranas, por lo cual habrá que pensar en esta enfermedad cuando nos encontremos con tumores renales con estas características en personas más jóvenes de lo esperado (8).
Histopatología
En general son tumores confinados al riñón (pT1/2). Las paredes de los quistes que lo constituyen son fibrosas (fig. 1) y están tapizadas por una o varias capas de células malignas. Éstas pueden estar ausentes, mostrando paredes desnudas o recubiertas de material fibrino-hemático. A veces las células malignas están aplanadas y su visualización puede ser dificultosa (fig. 2). Las áreas quísticas alternan con nidos sólidos (fig. 1) y en eso se diferencia del carcinoma de células claras variante multilocular, en el cual todo el tumor es típicamente macroquístico y sin áreas sólidas. Suele presentar cápsula fibrosa gruesa El patrón de crecimiento celular es el característico (fig. 3) mostrando también la peculiar distribución vascular capilar. Las células son poligonales y contienen un núcleo hipercromático, sin nucleolo aparente (fig. 4). El perfil inmunohistoquímico es similar a los carcinomas de células claras esporádicos y se especificará en los párrafos siguientes.
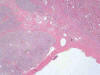
Fig. 1:
Imagen histológica panorámica de un carcinoma de células claras en el contexto
de la enfermedad de von Hippel-Lindau en el que se alternan áreas sólidas y
áreas quísticas (Hematoxilina-Eosina, x40).
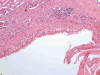
Fig. 2:
Pared quística fibrosa tapizada de forma discontinua por células claras asociada
a inflamación y depósito de fibrina en un caso asociado a enfermedad de von
Hippel-Lindau (Hematoxilina-Eosina, x100).
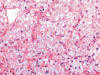
Fig. 3:
Carcinoma de células claras asociado a enfermedad de von Hippel-Lindau formando
nidos sólidos (Hematoxilina-Eosina, x250).
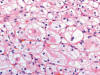
Fig. 4:
Detalle citológico de un carcinoma renal de células claras asociado a enfermedad
de von Hippel-Lindau mostrando bajo grado celular (Hematoxilina-Eosina, x400).
En definitiva, el carcinoma renal de células claras asociado a la enfermedad de von Hippel-Lindau es un carcinoma de células claras macroscópicamente sólido-quístico, multifocal y bilateral, de bajo grado histológico, que se desarrolla en personas más jóvenes de lo habitual y que tiene mejor pronóstico que el tipo convencional.
CARCINOMA RENAL DE CÉLULAS CLARAS ESPORÁDICO
Es el carcinoma renal más frecuente y su incidencia está aumentando en los últimos años (9,10). Aproximadamente el 70 a 75% de los cánceres renales son carcinomas de células claras esporádicos (11). Afecta más frecuentemente a varones de raza negra y en general por encima de los 40 años de edad. El hábito tabáquico y la obesidad, ésta última en mujeres, son factores predisponentes. Se postula que se originan a partir de la célula epitelial de ducto contorneado proximal (12).
Genética
Más del 90% de los carcinomas renales de células claras muestran la típica alteración citogenética en el brazo corto del cromosoma 3 (13), pudiendo encontrarse desde pérdidas completas del brazo hasta translocaciones o delecciones (14). De hecho, entre el 30% y el 56% de los carcinomas renales de células claras esporádicos tienen mutaciones en el gen de VHL y hasta el 19% tienen silenciamiento de mismo (15-17).
Macroscopía
Los carcinomas renales de células claras presentan una coloración amarillenta y un aspecto multilobulado (fig. 5), con áreas sólidas y quísticas, y con hemorragia y necrosis, circunstancias que normalmente acompañan a los casos de gran tamaño. En estos casos, la pieza quirúrgica remitida puede llegar a pesar varios kilos. Sin embargo, esta circunstancia es hoy en día excepcional. La mayor concienciación de la población frente a los síntomas, la estructura sanitaria y el elevado grado de refinamiento que han alcanzado las técnicas de imagen, hacen que cada vez más se detecten casos confinados al riñón y de pequeño tamaño. Esta circunstancia ha hecho posible el desarrollo en los últimos tiempos técnicas de crioablación o de ablación por radiofrecuencia por vía laparoscópica que minimizan los efectos de la cirugía convencional y que obtienen buenos resultados (18).
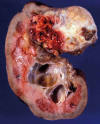
Fig. 5:
Imagen macroscópica de un carcinoma de células claras esporádico mostrando un
tumor de color amarillento con áreas hemorrágicas y con zonas centrales
cicatriciales blanquecinas. La tumoración se encuentra bien delimitada del
tejido renal adyacente.
Los tumores suelen estar bien delimitados del tejido renal adyacente por una cápsula fibrosa (fig. 6) que puede ser continua o no. Las áreas blanquecinas suelen estar relacionadas con la transformación sarcomatoide que a veces ocurre en este tumor. Las áreas de coloración parda suelen corresponder a zonas de hemorragia antigua. Alrededor del 15% de los casos tienen calcificaciones. La presencia de hueso metaplásico es muy poco frecuente.
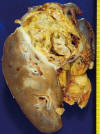
Fig. 6:
Imagen macroscópica de un carcinoma de células claras esporádico mostrando un
tumor sólido amarillento encapsulado y afectando focalmente la grasa perirenal.
Ocasionalmente, el carcinoma de células claras está constituido por quistes hasta el punto de ser confundido con lesiones no tumorales. Se trata de la variante multilocular, un subtipo de carcinoma de células claras raro y de bajo grado.
Histopatología
El carcinoma de células claras muestra una histología altamente característica (fig. 7). Las células del carcinoma de células claras muestran un citoplasma óptimamente vacío y una membrana citoplásmica muy bien definida (fig. 8). En ocasiones las células muestran citoplasmas débilmente eosinófilos en torno al núcleo (fig. 9), circunstancia observada en casos de alto grado nuclear. Si este cambio eosinofílico del citoplasma es muy extenso, pueden plantearse dudas diagnósticas iniciales con el carcinoma renal de células cromófobas. El núcleo puede mostrar morfologías variables y dependiendo de su morfología, de sus irregularidades y de la presencia de nucleolo (fig. 10), se ha establecido el sistema de Furhman (19) para determinar el grado. Esta forma de graduar los carcinomas renales está ampliamente extendida entre los patólogos de todo el mundo, aunque existen intentos recientes para modificar en parte esta forma de asignar el grado histológico a los carcinomas renales (20,21).
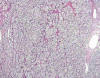
Fig. 7:
Imagen histológica panorámica de un carcinoma de células claras esporádico
mostrando el característico patrón de crecimiento sólido y en nidos con
vascularización finamente ramificada (Hematoxilina-Eosina, x40).
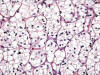
Fig. 8:
Detalle citológico de un carcinoma de células claras esporádico mostrando nidos
sólidos de células claras con bordes nítidos (Hematoxilina-Eosina, x400).
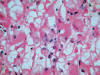
Fig. 9:
Detalle citológico de un carcinoma de células claras esporádico mostrando
células de citoplasma parcialmente eosinófilo (Hematoxilina-Eosina, x400).
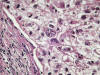
Fig. 10:
Detalle citológico de un carcinoma de células claras esporádico de alto grado
nuclear mostrando pleomorfismo nuclear marcado y nucleolos evidentes
(Hematoxilina-Eosina, x400).
Las células se disponen formando nidos, estructuras alveolares, cordones y papilas, o tapizando quistes de tamaño variable. La disposición papilar del tumor no debe ser interpretada como carcinoma papilar ya que estos casos mantienen las alteraciones genéticas localizadas en el cromosoma 3. El estroma contiene una red capilar muy prominente y típica. Las zonas centrales del tumor pueden mostrar áreas cicatriciales, esclerosis o edema. Es frecuente la hemorragia y la necrosis, sobre todo en los tumores grandes. Es típico encontrar acúmulos de hematíes en las luces de las estructuras alveolares (fig. 11). La transformación sarcomatoide puede producirse de forma brusca, aunque lo normal es que se puedan encontrar áreas de transición si se muestrea el tumor suficientemente. Estas áreas pueden presentar morfologías variadas, pero predomina el fenotipo fusocelular (fig. 12).
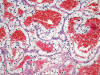
Fig. 11:
Carcinoma de células claras esporádico mostrando un patrón de crecimiento
alveolar con abundantes hematíes en la luz (Hematoxilina-Eosina, x250).
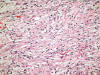
Fig. 12:
Carcinoma de células claras esporádico mostrando un patrón de crecimiento
fusocelular sarcomatoide (Hematoxilina-Eosina, x250).
La variante multilocular está constituida por quistes de tamaño variable, sin componente sólido acompañante (fig. 13). Los tabiques son de pared fina y están tapizados por células claras de morfología característica (fig. 14). La luz contiene un material acelular eosinófilo.
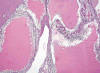
Fig. 13:
Imagen histológica panorámica de un carcinoma de células claras esporádico,
variedad multilocular, mostrando un crecimiento exclusivamente quístico
(Hematoxilina-Eosina, x40).
Fig. 14:
Detalle citológico de un carcinoma de células claras esporádico, variedad
multilocular, mostrando paredes finas tapizadas por células claras típicas de
bajo grado nuclear (Hematoxilina-Eosina, x400).
Inmunohistoquímica
Los carcinomas renales de células claras, independientemente de si son familiares, asociados a enfermedad de VHL o esporádicos, coexpresan citoqueratinas de bajo peso molecular (CKs 8, 18, 19) y vimentina (22). Esta doble tinción es característica, lo cual es un dato importante a la hora de valorar el origen tumoral en una metástasis a distancia de un primario desconocido. Asimismo, resulta positiva la inmunotinción con EMA, marcador de carcinoma renal (RCC), CD10 (fig. 15), CD13 (fig. 16), CD15 y MUC-1 (22). Como consecuencia de la alteración genética del cromosoma 3 que subyace en prácticamente todos los casos, la tinción con CA9 (fig. 17) y con ciclina D1 también es positiva.
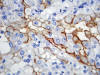
Fig. 15:
Positividad de membrana intensa para CD10 en un carcinoma de células claras
esporádico (CD10, contratinción con hematoxilina, x400).
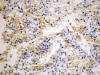
Fig. 16:
Positividad citoplásmica para CD13 en un carcinoma de células claras esporádico
(CD13, contratinción con hematoxilina, x250).

Fig. 17:
Positividad para anhidrasa carbónica 9 (CA9) en un carcinoma de células claras
esporádico (CA9, contratinción con hematoxilina, x250).
Ocasionalmente pueden ser positivas las tinciones con CEA, a-fetoproteína, proteína S-100, a-1-antitripisina y fosfatasa alcalina placentaria. Resultan negativas la CK7, CK14, 34betaE12, CK20, parvalbúmina, CD117, racemasa (p504S) y la e-cadherina (22).
CARCINOMAS RENALES DE CÉLULAS CLARAS DE TIPO FAMILIAR (NO ASOCIADO A ENFERMEDAD DE VON HIPPEL-LINDAU)
Existen carcinomas renales de células claras de agrupación familiar, aunque son extraordinariamente infrecuentes. Histológicamente todos ellos son indistinguibles del esporádico.
Carcinoma renal de células claras familiar asociado a translocación del cromosoma 3
Se trata de carcinomas renales causados por diversos puntos de rotura en el cromosoma 3 y son típicamente múltiples y bilaterales. La primera translocación balanceada t(3;8)(p14;q24) se describió en 1979 (23) y desde entonces se han descrito varias más (24), todas ellas afectando al brazo corto del cromosoma 3. Los casos descritos en hermanos gemelos en 1979 mostraban predisposición a desarrollar carcinoma renal de células claras bilaterales y carcinoma tiroideo (23). El punto de corte correspondía, en el cromosoma 3 con el gen FHIT (triada de histidina frágil) (25) y en el cromosoma 8 con el gen TRC8 (26). Ambos funcionan como genes supresores y la translocación los inactiva. El FHIT está situado en la región frágil del genoma humano –FRA3B– y el TRC8 es homólogo del que en la mosca Drosophila codirige la embriogénesis. Éste último actúa siguiendo pasos comunes al gen de VHL (26).
El hecho de albergar al gen VHL y de haberse descrito varios puntos de rotura diferentes al comentado aquí en el brazo corto del cromosoma 3 lleva a pensar que esta parte del genoma humano contiene varios genes implicados en el desarrollo del cáncer renal.
Carcinoma renal de células claras familiar
Se define como tal a aquellos casos de carcinoma renal de células claras que afecta a dos o más familiares en primer grado y, a diferencia del los casos del grupo anterior, tienden a ser tumores únicos y confinados al riñón. Existen muy pocos casos (27). No se ha detectado aún la mutación responsable, estando ausentes las mutaciones en línea germinal del gen VHL y todo tipo de translocaciones detectables en el cromosoma 3.
CARCINOMAS RENALES CON TRANSLOCACIÓN
Carcinoma renal con traslocación Xp11.2
Con este nombre se agrupan una serie de carcinomas renales que se presentan en la edad pediátrica y que tienen en común una translocación en la que el punto de ruptura está en Xp11.2. Como consecuencia de ello se producen fusiones del factor de transcripción TFE3, que es el que se codifica precisamente en esa región cromosómica, con otros genes (28,29). Hasta la fecha se han descrito 5 diferentes fusiones que afectan al TFE3 (30-32) (tabla 1). Una de ellas, la ASPL-TFE3, es idéntica a la que se produce en el sarcoma alveolar de partes blandas (31). Muchos de estos pacientes se caracterizan por haber recibido, varios años antes, tratamientos de quimioterapia citotóxica por leucemias y otros tumores, e incluso por lupus eritematoso sistémico (33).

Los carcinomas renales con translocación Xp11.2 tienen una morfología típica. El examen macroscópico es superponible al carcinoma de células claras convencional. Se trata de carcinomas compuestos por células claras de gran tamaño (fig. 18) que forman nidos, alvéolos y papilas (fig. 19) acompañados de abundantes cuerpos de psammoma. A veces presentan citoplasmas levemente eosinófilos (fig. 20). Todos estos carcinomas, antes de ser evaluados e identificados genéticamente, pertenecían al raro grupo de carcinomas renales de la infancia y adolescencia. Este dato es muy útil a la hora de rescatar posibles nuevos casos de nuestros archivos. Así, Chian-Garcia y cols (34), Atinok y cols (35) y Bruder y cols (36) encuentran que el 41%, el 75% y el 20%, respectivamente, de los carcinomas renales de sus archivos pertenecientes a este rango de edad eran carcinomas con translocación Xp11.2. De forma característica, estos tumores son positivos con los marcadores inmunohistoquímicos del melanoma (Melan A y HMB45) y no expresan, o lo hacen muy débilmente, citoqueratinas y vimentina (28,29). También se detecta positividad con CD10 y con el marcador del carcinoma renal (fig. 21). Más específicamente, este grupo de carcinomas muestra positividad nuclear con el factor de transcripción TFE3 en inmunohistoquímica de material incluido en parafina (fig. 22).
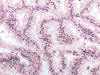
Fig. 18:
Imagen característica de un carcinoma renal con translocación Xp11.2 y fusión
ASPL-TFE3 mostrando papilas tapizadas por células claras de talla alta
(Hematoxilina-Eosina, x250).
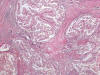
Fig. 19:
Imagen histológica panorámica de un carcinoma renal con translocación Xp11.2 y
fusión ASPL-TFE3 mostrando un crecimiento en nidos inmersos en un estroma
fibroso (Hematoxilina-Eosina, x40).
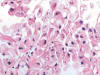
Fig. 20:
Detalle citológico de un carcinoma renal con translocación Xp11.2 y fusión
ASPL-TFE3 mostrando células de talla grande, con citoplasmas parcialmente
eosinófilos, y pleomorfismo nuclear marcado (Hematoxilina-Eosina, x400).
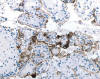
Fig. 21:
Positividad para CD10 en un carcinoma renal con translocación Xp11.2 y fusión
ASPL-TFE3 (CD10, contratinción con hematoxilina, x250).
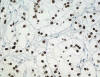
Fig. 22:
Positividad para TFE3 en un carcinoma renal con translocación Xp11.2 y fusión
ASPL-TFE3 (TFE3, contratinción con hematoxilina, x250).
El carcinoma renal con fusión ASPL-TFE3 [t(X,17) (p11.2;q25)] se presenta en estadios avanzados y en pacientes por debajo de los 18 años de edad. Casi todos los casos descritos muestran metástasis ganglionares a pesar del pequeño tamaño del tumor primario. A pesar de ello, muestran un curso clínico indolente. Muestran abundantes nódulos hialinos y cuerpos de psammoma (fig. 23), citoplasmas claros muy voluminosos, cromatina nuclear vesicular y nucleolo prominente. Las células son poco cohesivas, lo cual produce imágenes alveolares y pseudopapilares. Al microscopio electrónico estos carcinomas contienen gránulos citoplásmicos rodeados por membrana y cristales romboideos similares a los que se detectan en el sarcoma alveolar de partes blandas (31).
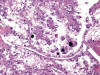
Fig. 23:
Imagen histológica de un carcinoma renal con translocación Xp11.2 y fusión
PRCC-TFE3 mostrando células claras, papilas y cuerpos de psammoma
(Hematoxilina-Eosina, x100).
El carcinoma renal con fusión PRCC-TFE3 [t(X;1) (p11.2;q21) presenta una arquitectura más compacta, en nidos, no tan papilar y citoplasmas menos abundantes (fig. 24), menos cuerpos de psammoma y menos nódulos hialinos (32). Pueden presentar una pseudocápsula fibrosa calcificada identificable incluso desde el punto de vista macroscópico. Ultraestructuralmente, las células presentan microtúbulos similares al los descritos en el melanoma y el condrosarcoma mixoide extraesquelético (32).
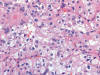
Fig. 24:
Detalle citológico de un carcinoma renal con translocación Xp11.2 y fusión
PRCC-TFE3 mostrando células claras con citoplasma escaso, parcialmente
eosinófilo, creciendo en forma de nidos sólidos (Hematoxilina-Eosina, x250).
El resto de carcinomas renales con translocación xp11.2 (PSF-TFE3, NonO-TFE3, CLTC-TFE3) no han sido aún caracterizados debido a la escasez de casos descritos.
Carcinoma renal con t(6;11)(p21;q12)
El primer caso de carcinoma renal con esta translocación específica se describió en 1996 (37) y desde entonces se han descrito 9 casos (38). Las características clínicas y patológicas de este tipo de carcinomas renal se han descrito recientemente (39).
La t(6;11) fusiona el gen alfa en 11q12, un gen de función desconocida, con el primer intrón del factor de transcripción TFEB en 6p21 (40). El punto de rotura en TFEB mantiene completa la región codificante de TFEB en la fusión.
Histológicamente, se trata de neoplasias sólidas, sin estructuras papilares, compuestas por nidos y túbulos de células epitelioides poligonales acompañados de una fina y abundante trama capilar. Las células tienen citoplasmas claros o granulares eosinofílicos, con bordes citoplásmicos bien definidos, núcleos redondeados y nucleolos pequeños. Se observa también abundantes nódulos de material de membrana basal. Las mitosis son escasas.
Por inmunohistoquímica, estos tumores son negativos para citoqueratinas y positivos para algunos marcadores de melanoma como HMB45 y Melan-A y negativos para otros como el factor de transcripción de microftalmia. Asimismo, es específica de este carcinoma la positividad nuclear para TFEB.
CARCINOMA PAPILAR RENAL HEREDITARIO
Clínica
El carcinoma renal papilar hereditario es una enfermedad autosómica dominante que se caracteriza por tumores bilaterales múltiples de comienzo tardío, aunque existen casos aislados de aparición temprana (41). La enfermedad muestra un curso indolente. Muchos pacientes están asintomáticos al diagnóstico, y son descubiertos en exámenes radiológicos rutinarios o realizados por otras causas. Radiológicamente son tumores hipovasculares y su imagen varía poco tras la administración de contraste (42).
Histología
La enfermedad se caracteriza por múltiples tumores de morfología papilar o túbulo-papilar. Se observa toda la gama, desde pequeños grupos microscópicos y adenomas papilares, hasta carcinomas papilares de tipo 1 (43). Las células malignas muestran bajo grado nuclear. El patrón histológico tubular y papilar puede presentar áreas sólidas semejando al adenoma metanéfrico. Los ejes papilares son finos y contienen, al igual que los carcinomas papilares esporádicos, acúmulos de histiocitos espumosos y cuerpos de psammoma.
Curiosamente, hasta el 70% de los carcinomas papilares hereditarios contienen células claras (43), en especial en aquellas áreas con necrosis o hemorragia. Las células claras muestran un citoplasma a veces espumoso y un núcleo cromático, con nucleolo basófilo. El carácter eminentemente papilar de la neoplasia, junto a la ausencia del patrón vascular típico, ayuda a distinguir el carcinoma papilar renal hereditario con células claras del carcinoma renal de células claras. El resto del diagnóstico diferencial incluye al resto de carcinomas papilares, al carcinoma de los túbulos colectores con predominio papilar y, aquellos casos con un crecimiento preferentemente sólido, con el adenoma metanéfrico. Este último carece de las alteraciones genéticas propias del carcinoma papilar (44,45).
Genética
El carcinoma renal papilar hereditario está causado por mutaciones en el proto-oncogén c-MET, que está constituido por 20 exones y se localiza en 7q31-34 (46,47). Codifica una tirosin-quinasa que está implicada en diversas respuestas biológicas relacionadas con el control del crecimiento celular, la supervivencia celular y la morfogénesis (48). c-MET y su ligando, el factor de crecimiento hepatocitario, son necesarios para el desarrollo embrionario normal del músculo y del hígado y su ausencia es incompatible con la vida (49). La sobre-expresión de c-MET, al igual que la de sus proto-oncogenes análogos c-kit y RET, está asociado a múltiples neoplasias (50). Las alteraciones responsables del carcinoma renal papilar hereditario suelen ser bien mutaciones en línea germinal a nivel de los exones 16 a 19, o bien trisomías 7. Un pequeño subgrupo de carcinomas renales papilares esporádicos también presenta mutaciones de c-MET idénticas a las de las formas hereditarias (43,44).
CARCINOMA RENAL ASOCIADO A ESCLEROSIS TUBEROSA
Genética
La esclerosis tuberosa es un síndrome caracterizado por alteraciones en el complejo esclerosis tuberosa que es un conjunto de genes supresores tumorales y que lleva a retraso mental, autismo, crisis epilépticas y tumores cerebrales, retinianos, renales, cardiacos y cutáneos. Es un defecto genético que se transmite de manera autosómica dominante con un 95% de penetrancia y que afecta a 1 de cada 11.000 nacimientos (51).
La enfermedad se basa en mutaciones en línea germinal bien en el gen TSC1 (9q34)(52) o en el TSC2 (16p13) (53) y sigue el patrón de Knudson de enfermedad genética desarrollada en dos «hits», en el que un alelo se inactiva en línea germinal y el otro lo hace mediante mutación somática o por pérdida de heterozigosidad (54).
TSC1 codifica la hamartina y TSC2 la tuberina, cuyas funciones han sido reconocidas recientemente (55). El complejo TSC1/TSC2 actúa a varios niveles, inhibiendo mTOR, una kinasa que regula la síntesis proteica y regulando diversos factores implicados en el crecimiento celular tanto en células mesenquimales como epiteliales (56), lo que convierte a la esclerosis tuberosa en un modelo único de génesis tumoral. El complejo TSC1/TSC2 regula tanto la proliferación como la diferenciación de la célula renal precursora que deriva del mesodermo embrionario (57), el cual tiene capacidades de para desarrollar elementos epiteliales y estromales. La alteración en este complejo explica el desarrollo tanto de angiomiolipomas como de oncocitomas o carcinomas en esta enfermedad y justifica la existencia de angiomiolipomas con componente epitelial como se ha descrito recientemente (58) (fig. 25).
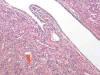
Fig. 25:
Imagen histológica de un angiomiolipoma renal conteniendo en su espesor quistes
epiteliales (Hematoxilina-Eosina, x100).
Clínica
Los tumores renales más frecuentes en este síndrome son los angiomiolipomas (80%), pero también pueden desarrollarse neoplasias epiteliales como oncocitomas y carcinomas de células renales (5%). En la esclerosis tuberosa, los carcinomas renales se producen en la juventud e incluso antes, en la infancia (56).
Histopatología
El carcinoma renal asociado a esclerosis tuberosa se caracteriza por su heterogeneidad, pudiendo encontrarse carcinomas con células claras, papilares y cromófobos (59). Esta heterogeneidad del carcinoma renal asociado a esclerosis tuberosa contrasta con la homogeneidad descrita en las alteraciones del VHL en el que el carcinoma siempre es de célula clara, o en las relacionadas con el gen MET, que son carcinomas papilares.
La imagen histológica y el patrón inmunohistoquímico son totalmente superponibles a los del carcinoma renal de células claras, tipo esporádico.
CARCINOMA RENAL ASOCIADO AL SÍNDROME DE BIRT-HOGG-DUBÉ
El síndrome de Birt-Hogg-Dubé es una enfermedad autosómica dominante que consiste en una triada de lesiones cutáneas compuesta por fibrofoliculomas, tricodiscomas, y acrocordones (60), al que se le añaden con cierta frecuencia neumotórax espontáneos, neoplasias colorrectales y diversos tipos de carcinomas renales entre los que predominan los carcinomas de células cromófobas (61). Aproximadamente el 9% de los carcinomas renales desarrollados en este síndrome son carcinomas de células claras indistinguibles histológicamente de las variedades esporádicas (62).
La alteración genética en este síndrome se localiza en el cromosoma 17 (17p12-q12.2) (63). Los análisis moleculares de los carcinomas de células claras desarrollados en esta enfermedad muestran pérdidas en 3p y mutaciones en el gen de la enfermedad de von-Hippel-Lindau (62). No obstante, estos casos poco frecuentes de carcinomas de células claras en el contexto de este síndrome pueden no ser manifestaciones genuinas del mismo, como ha sido previamente mencionado por Pavlovich y cols (62). De esta manera, la relación entre el carcinoma renal de células claras y el síndrome de Birt-Hogg-Dubé está hoy en día en entredicho.
CARCINOMA RENAL DE CÉLULAS CROMÓFOBAS
Esta variedad de carcinoma renal fue descrita por Thoenes en 1985 (64). Se han descrito dos subtipos histológicos, la variedad clásica y la eosinofílica, con un mejor pronóstico según algunos autores (65) para esta última. El tumor se origina en la neurona inferior y se ha relacionado histogenéticamente con los oncocitomas renales.
Hoy en día se considera que el carcinoma de células cromófobas y el oncocitoma renal representan los dos extremos de un espectro de tumores oncocíticos con múltiples estadios intermedios, y que muchos de ellos muestran histologías mixtas o híbridas que representan un cierto grado mal conocido de progresión de un tumor hacia el otro (66-68).
Genética
El carcinoma de células cromófobas presenta múltiples y complejas pérdidas cromosómicas que afectan a los cromosomas Y, 1, 2, 6, 10, 13, 17 y 21 (67).
Histopatología
Al microscopio, la variedad clásica está compuesta por células grandes de contorno poliédrico, con membrana citoplásmica evidente y citoplasma finamente granular o claro, dispuestas en nidos, cordones, o ductos, que pueden ser confundidas con un carcinoma renal de células claras que contenga células granulares, hecho que ocurre con cierta frecuencia. Los oncocitomas y el resto de tumores oncocíticos, incluyendo la variedad eosinofílica del carcinoma de células cromófobas, tienen un citoplasma intensamente eosinofílico y granular y normalmente no plantean problemas diagnósticos. Sin embargo, se ha visto que las células de estos tumores, cuando están localizadas en la cicatriz central, pueden mostrar cierto aclaramiento del citoplasma y este hecho puede plantear eventualmente cierta confusión.
El perfil inmunohistoquímico más común en el carcinoma de células cromófobas incluye positividad para CK7, EMA, MUC1, e-cadherina, c-kit y parvalbúmina. Resultan negativos CK20, marcador de carcinoma renal (RCC marker), CD10, vimentina, racemasa (p504S) y queratinas de alto peso molecular (22).
CARCINOMA RENAL ASOCIADO AENFERMEDAD QUÍSTICA RENAL
Los pacientes con enfermedad quística renal muestran una incidencia elevada de carcinoma renal asociado, particularmente aquellos que desarrollan enfermedad quística adquirida (69). Aunque la diálisis es la causa más común (70), el rechazo crónico del transplante también puede ser causa de enfermedad quística adquirida (71). Los tumores que se desarrollan en estos contextos ocurren en pacientes más jóvenes, tienen una mayor tendencia a la multicentricidad y bilateralidad, y siguen un curso clínico menos agresivo que las variedades esporádicas de carcinoma renal, siendo en muchos casos asintomáticos (69).
Genética
No se han descrito alteraciones genéticas específicas en este grupo de tumores. Cossu-Rocca y cols (72) han encontrado recientemente ganancias cromosómicas variadas en los cromosomas 1, 2, 6 y 10 en dos de sus tres casos publicados.
Histopatología
Según ha sido recientemente publicado por Tickoo y cols (73), estas neoplasias pueden ser, bien histológicamente similares en todo a las formas esporádicas de carcinoma renal (células claras, papilar, células cromófobas), o bien pueden ser peculiares y específicas de la enfermedad. En este último grupo se pueden distinguir dos tipos diferentes de histologías.
El primer grupo de tumores, denominado carcinoma renal asociado a enfermedad quística renal, muestra focos de crecimiento papilar, áreas cribiformes compuestas por luces y puentes rígidos, y células eosinofílicas con nucleolos prominentes (grado 3 de Fuhrman). Se pueden identificar cristales e oxalato en el seno del tumor. En estos tumores se detecta positividad para racemasa (p504S) y parvalbúmina. La CK7 es negativa (73).
El segundo grupo, denominado carcinoma renal papilar de células claras del riñón en estadio terminal, está compuesto por tumores predominantemente quísticos constituidos por papilas tapizadas casi exclusivamente por células claras de bajo grado. Estos tumores son positivos con CK7 y negativos con racemasa (p504S) (73).
CONCLUSIONES
La clasificación de los tumores renales de la OMS publicada en 2004 reconoce 49 tipos diferentes de neoplasias renales (74). Hasta la fecha, las clasificaciones de los tumores renales se basaban fundamentalmente en parámetros histológicos. Sin embargo, como se ha podido demostrar en parte en esta revisión, la morfología sola es insuficiente para clasificar los tumores renales. En este sentido son varios los autores que demuestran la utilidad de clasificar los tumores renales en base a su perfil molecular (75).
Los carcinomas renales de células claras son, con mucho, los tumores más frecuentes en este órgano y cualquier patólogo se enfrenta a ellos con relativa frecuencia. Sin embargo, no todos ellos son la misma enfermedad. Debajo de una morfología muy similar subyacen varias enfermedades que responden a alteraciones genéticas totalmente específicas. El conocimiento de todas estas posibilidades diagnósticas es importante porque, en muchos casos, la combinación de datos clínicos y morfológicos en la hematoxilina-eosina es suficiente para sospechar entidades tan infrecuentes como los carcinomas con translocación. Esta revisión pretende precisamente eso.
BIBLIOGRAFÍA
Fletcher CDM, editor. Diagnostic Histopathology of Tumors. 2nd Edition. London: Churchill Livingstone; 2000.
Rosai J, editor. Rosai and Ackerman’s Surgical Pathology. 9th Edition. Edinburgh: Mosby; 2004.
Maher ER, Kaelin WG Jr. von Hippel-Lindau disease. Medicine (Bal) 1997; 76: 381-91.
Seizinger BR, Rouleau GA, Ozelius LJ, et al. von Hippel-Lindau disease maps to the region of chromosome 3 associated with renal cell carcinoma. Nature 1988; 332: 268-9.
Zbar B, Kishida T, Chen F, et al. Germline mutations in the von Hippel-Lindau disease (VHL) gene in families from North America, Europe and Japan. Hum Mutat 1996; 8: 348-57.
Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol 2004; 22: 4991-5004.
Sufan RI, Jewett MAS, Ohh M. The role of von Hippel-Lindau tumor suppressor protein and hypoxia in renal clear cell carcinoma. Am J Physiol Renal Physiol 2004; 287: 1-6.
Grignon DJ, Che M. Clear cell renal cell carcinoma. Clin Lab Med 2005; 25: 305-16.
Jemal A, Siegel R, Ward E, et al. Cancer statistics, 2007. CA Cancer J Clin 2007; 57: 43-66.
Chow WH, Devesa SS, Warren JL, et al. Rising incidenceof renal cell cancer in the United States. JAMA 1999; 281: 1628-31.
Thoenes W, Störkel S, Rumpelt HJ. Histopathology and classification of renal cell tumors (adenomas, oncocytomas, and carcinomas). Path Res Pract 1986; 181:125-43.
Yoshida SO, Imam A, Olson CA, et al. Proximal renal tubular surface membrane antigens identified in primary and metastatic renal cell carcinomas. Arch Pathol Lab Med 1986; 110: 825-32.
Anglard P, Troy K, Brauch H, et al Molecular analysis of genetic changes in the origin and development of renal cell carcinoma. Cancer Res 1991; 51: 1071-7.
Yoshida MA, Ohyashiki K, Ochi H, et al. Cytogenetic studies of tumor tissue from patients with nonfamilial renal cell carcinoma. Cancer Res 1986; 46: 2139-47.
Foster K, Prowse A, van der Berg A, et al. Somatic mutations of the von Hippel-Lindau disease tumour suppressor gene in non-familial clear cell renal carcinoma. Hum Mol Genet 1994; 3: 2169-73.
Schraml P, Struckmann K, Hatz F, et al. VHL mutations and their correlation with tumour cell proliferation, microvessel density, and patient prognosis in clear cell renal cell carcinoma. J Pathol 2002; 196: 186-93.
Bottaro DP, Linehan WM. Multifocal renal cancer: genetic basis and its medical relevance. Clin Cancer Res 2005; 11: 7206-8.
Park S, Cadeddu JA, Shingleton WB. Oncologic outcomes for ablative therapy of kidney cancer. Curr Urol Rep 2007; 8: 31-7.
Fuhrman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma. Am J Surg Pathol 1982; 6: 655-63.
Novara G, Martignoni G, Artibani W, Ficarra V. Grading systems in renal cell carcinoma. J Urol 2007; 177: 430-6.
Rioux-Leclercq N, Karakiewicz PI, Trinh QD, et al. Prognostic ability of simplified nuclear grading on renal cell carcinoma. Cancer 2007; doi: 10.1002/cncr.22463.
Zhou M, Roma A, Magi-Galluzzi C. The usefulness of immunohistochemical markers in the diferencial diagnosis of renal neoplasms. Clin Lab Med 2005; 25: 247-57.
Cohen AJ, Li FP, Berg S, et al. Hereditary renal cell carcinoma associated with a chromosomal translocation. N Eng J Med 1979; 301: 592-5.
van Kessel AG, Wijnhoven H, Bodmer D, et al. Renal cell cancer: chromosome 3 translocations and risk factors. J Natl Cancer Inst 1999; 91: 1159-60.
Ohta M, Inoue H, Cotticelli MG, et al. The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal cell carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 1996; 84: 587-97.
Gemmill RM, Bemis LT, Lee JP, et al. The TRC8 hereditary kidney cancer gene suppresses growth and functions with VHL common pathway. Oncogene 2002; 21: 3507-16.
Teh BT, Giraud S, Sari NF, et al. Familial non-VHL non-papillary clear-cell renal cancer. Lancet 1997; 349: 848-9.
Argani P, Ladanyi M. Translocation carcinomas of the kidney. Clin Lab Med 2005; 25: 363-78.
Argani P, Ladanyi M. The evolving story of renal translocation carcinomas. Am J Clin Pathol 2006; 126: 332-4.
Clark J, Lu YL, Sidhar SK, et al. Fusion of splicing factor genes PSF and NonO (p54nrb) to the TFE3 gene in papillary renal cell carcinoma. Oncogene 1997; 15: 2233-9.
Argani P, Antonescu CR, Illei PB, et al. Primary renal neoplasms with the ASLP-TFE3 gene fusion of alveolar sofá part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents Am J Pathol 2001; 159: 179-92.
Argani P, Antonescu CR, Couturier J, et al. PRCC-TFE3 renal cell carcinomas: morphologic, immunohistochemical, ultrastructural and molecular analysis o fan entity associated with the t(X;1)(p11.2;q21). Am J Surg Pathol 2002; 26: 1553-66.
Argani P Lae M, Ballard ET, et al. Translocation carcinomas of the kidney after chemotherapy in childwood. J Clin Oncol 2006; 24: 1529-34.
Chian-Garcia CA, Torres-Cabala CA, Eyler R, et al. Renal cell carcinoma in children and young adults: a clinicopathologic and immunohistochemical study of 14 cases. Mod Pathol 2003; 16: 145A.
Altinok G, Kattar MM, Mohamed A, et al. Pediatric renal carcinoma associated with Xp11.2 translocations/TFE3 gene fusions and clinicopathologic associations. Pediatr Dev Pathol 2005; 8: 168-80.
Bruder E, Passera O, Harms D, et al. Morphologic and molecular characterization of renal cell carcinoma in children and young adults. Am J Surg Pathol 2004; 28: 1117-32.
Dijkhuizen T, ven den Berg E, Störkel S, et al. Two cases of renal cell carcinoma, clear cell type, revealing t(6;11)(p21;q12)(abstract) Cancer Genet Cytogenet 1996; 91: 141.
38. Argani P, Lae M, Hutchison B, et al. Renal carcinomas with the t(6;11)(p21;q12). Clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol 2005; 29: 230-40.
Argani P, Hawkins A, Griffin CA, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol 2001; 158: 2089-96.
Davis IJ, His BL, Arroyo JD, et al. Cloning of a novel alpha-TFEB fusion in renal tumors harbouring the t(6;11)(p21;q12) chromosome translocation. Proc Natl Acad Sci USA 2003; 100: 6051-6.
Schmidt LS, Nickerson ML, Angeloni D, et al. Early onset hereditary papillary renal carcinoma: germline missense mutations in the tyrosine kinase domain of the met proto-oncogene. J Urol 2004; 172: 1256-61.
Choyke P, Walther M, Glenn G, et al. Imaging features of hereditary papillary renal cancers. J Comput Assist Tomogr 1997; 21: 737-741.
Lubensky IA, Schmidt L, Zhuang Z. Hereditary and sporadic papillary renal cell carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol 1999; 155: 517-26.
Schmidt L, Junker K, Nakaigawa N, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 18: 1999; 2343-50.
Brunelli M, Eble JN, Zhang S, et al. Metanephric adenoma lacks the gains of chromosomes 7 and 17 and loss of Y that are typical of papillary renal cell carcinoma and papillary adenoma. Mod Pathol 2003; 16: 1060-3.
Schmidt L, Duh FM, Chen F, et al. Germline and somatic mutationsi n the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997; 16: 68-73.
Bottaro DP, Linehan WM. Multifocal renal cancer: genetic basis and its medical relevance. Clin Cancer Res 2005; 11: 7206-8.
Cohen D, Zhou M. Molecular genetics of familial renal cell carcinomas syndromes. Clin Lab Med 2005; 25: 259-77.
Ponzetto C, Pante G, Prunotto C, et al. Met signalling mutants as tools for developmental studies. Int J Dev Biol 2000; 44: 645-53.
Di Renzo MF, Narsimhan RP, Olivero M, et al. Expression of the Met/HGF receptor in normal and neoplastic human tisúes. Oncogene 1991; 6: 1997-2003.
O’Callaghan FJK, Shiell AW, Osborne JP, et al. Prevalence of tuberous sclerosis estimated by capture-recapture analysis. Lancet 1998; 351: 1490.
van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277: 805-8.
European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993; 75: 1305-15.
Bjornsson J, Short MP, Kwiatkowski DJ, et al. Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am J Pathol 1996; 149: 1201-8.
Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004; 23: 3151-71.
Robertson FM, Cendron M, Klauber GT, et al. Renal cell carcinoma in association with tuberous sclerosis in children. J Pediatr Surg 1996; 31: 729-30.
Henske EP. Tuberous sclerosis and the kidney: from mesenchyme to epithelium, and beyond. Pediatr Nephrol 2005; 20: 854-7.
Fine SW, Reuter VE, Epstein JI, et al. Angiomyolipoma with epithelial cysts (AMLEC). A distinct cystic variant of angiomyolipoma. Am J Surg Pathol 2006; 30: 593-9.
Al-Saleem T, Wessner LL, Scheithauer BW, et al. Malignant tumors of the kidney, brain and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer 1998; 83: 2208-16.
Birt A, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Ddermatol 1977; 113: 1674-7.
Khoo SK, Kahnoski K, Sugimura J, et al. Inactivation of BHD in sparadic renal tumors. Cancer Res 2003; 63: 4583-7.
Pavlovich CP, Walther MM; Eyler RA, et al. Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 2002; 26: 1542-52.
Khoo SK, Bradley M, Wong FK, et al. Birt-Hogg-Dubé syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene 2001; 20: 5239-42.
Thoenes W, Storkel S, Rumpelt HJ. Human chromophobe renal cell carcinoma. Virchow´s Arch (B Cell Pathol) 1985; 48: 207-17.
Onishi T, Oishi Y, Yanada S, Abe K, Hasegawa T, Maeda S. Prognostic implications of histological features in patients with chromophobe cell renal carcinoma. BJU Int 2002; 90: 529-32.
Al-Saleem T, Cairns P, Dulaimi EA, Feder M, Testa JR, Uzzo RG. The genetics of renal oncocytosis: a possible model for neoplastic progression. Cancer Genet Cytogenet 2004; 152: 23-8.
Abrahams NA, Tamboli P. Oncocytic renal neoplasms: diagnostic considerations. Clin Lab Med 2005; 25: 317-39.
Salido M, Lloreta J, Melero C, et al. Insertion (8;11) in a renal oncocytoma with multifocal transformation to chromophobe renal cell carcinoma. Cancer Genet Cytogenet 2005; 163: 160-3.
Truong LD, Choi YJ, Shen SS, Ayala G, Amato R, Krishnan B. Renal cystic neoplasms and renal neoplasms associated with cystic renal diseases: pathogenetic and molecular links. Adv Anat Pathol 2003; 10: 135-59.
Kojima Y, Takahara S, Miyake O, Nonomura N, Morimoto A, Mori H. Renal cell carcinoma in dialysis patients: a single center experience. Int J Urol 2006; 13: 1045-8.
Pope JC, Koch MO, Bluth RF. Renal cell carcinoma in patients with end-stage renal disease: a comparison of clinical significance in patients receiving hemodialysis and those with renal transplants. Urology 1994; 44: 497-501.
Cossu-Rocca P, Eble JN, Zhang S, Martignoni G, Brunelli M, Cheng L. Acquired cystic disease-associated renal tumors: an immunohistochemical and fluorescence in situ hybridization study. Mod Pathol 2006; 19: 780-7.
Tickoo SK, dePeralta-Venturina MN, Harik LR, et al. Spectrum of epithelial neoplasms in end-stage renal disease. An experience from 66 tumor-bearing kidneys with emphasis on histologic patterns distinct from those in sporadic adult renal neoplasia. Am J Surg Pathol 2006; 30: 141-53.
Eble JN, Sauter G, Epstein JI, et al. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Lyon: IARC Press; 2004.
Yang XJ, Sugimura J, Schafernak KT, et al. Classification of renal neoplasms based on molecular signatures. J Urol 2006; 175: 2302-6.
![]()