
Vol. 41, n.º 2, 2008
|
REVISTA
ESPAÑOLA DE
Vol. 41, n.º 2, 2008 |
REVISIONES
José I. López1, Marta Saiz Ruiz de Loizaga1, Cristina Magi-Galluzzi2
1 Departamento de
Anatomía Patológica. Hospital de Cruces. Universidad del País Vasco (EHU/UPV).
Barakaldo, Bizkaia.
2 Department of Pathology. Cleveland Clinic. Cleveland, Ohio, USA.
magic@ccf.org
joseignacio.lopez@osakidetza.net
RESUMEN
En este trabajo se revisa el amplio grupo de carcinomas renales constituidos por papilas, un hecho relativamente frecuente en la práctica diaria en nuestro medio. Este abordaje es pertinente debido a que no todos los carcinomas renales constituidos por papilas son auténticos carcinomas renales papilares. Dicha aproximación puramente morfológica no reniega de la nueva clasificación de la OMS vigente, sino que la complementa. El trabajo está dirigido más al patólogo general que se enfrenta a casos muy variados con alta presión asistencial, que al subespecialista acostumbrado a profundizar en casos complejos y con grandes medios técnicos en el campo del cáncer renal. Se revisan las características clínicas, histológicas, inmunohistoquímicas y genéticas del carcinoma renal papilar esporádico, del carcinoma renal papilar hereditario, del carcinoma renal asociado a leiomiomatosis hereditaria, del carcinoma renal papilar asociado a carcinoma papilar tiroideo hereditario, y de algunos carcinomas asociados bien a la enfermedad de Birt-Hogg-Dubé, o a translocaciones Xp11.2.
Palabras clave: Carcinoma renal papilar, alteración genética, diagnóstico, diagnóstico diferencial.
SUMMARY
This paper reviews a wide range of renal cell carcinomas showing papillary architecture, a relatively common finding in our daily practice. This approach to the problem is useful due to the fact that not all renal cell carcinomas composed of papillae are true papillary renal cell carcinomas. The purely morphological approach complements rather than denies the current WHO classification. This review targets general pathologists who face a wide variety of cases under high pressure to provide patient care, rather than sub-specialists in the field of renal cancer who are used to in-depth studies of complicated cases and who have important technical means available to them. The clinical, histological, immunohistochemical and genetic features of sporadic papillary renal cell carcinoma, hereditary papillary renal cell carcinoma, renal cell carcinoma associated with hereditary leiomyomatosis, papillary renal cell carcinoma associated with hereditary thyroid papillary carcinoma, and some carcinomas associated with Birt-Hogg-Dubé syndrome or to Xp11.2 translocations are reviewed.
Keywords: Papillary renal cell carcinoma, genetic alteration, diagnosis, differential diagnosis.
INTRODUCCIÓN
La arquitectura papilar es muy común en patología, apareciendo prácticamente todos los días tanto en histología normal, como en tumores benignos y en neoplasias malignas. Morfológicamente, se distinguen varios tipos de estructuras papilares. La papila verdadera consta de un eje fibrovascular más o menos evidente, y más o menos complejo o ramificado, sobre el que se aponen células de todo tipo y condición. Por el contrario, la pseudopapila, otra estructura frecuente en histopatología, carece de eje conectivo vascular y las células se aponen sobre las del lado contrario mediando entre ellas solamente la membrana basal. A medio camino entre ambas se sitúa la micropapila, estructura fina, delicada y delgada con ejes poco evidentes. A veces éstas últimas presentan morfologías mixtas, con partes proximales conteniendo ejes, y partes distales sin él. Todas pueden aparecer en una amplia gama de tumores benignos y malignos. Bastará echar un vistazo al índice alfabético de cualquier libro de patología quirúrgica al uso (1,2) para darse cuenta de cuán larga y variada puede ser la lista.
En ocasiones, la papila, la pseudopapila, o la micropapila, confieren al tumor un carácter exclusivo, definitorio, que lo convierte en entidad propia con pronóstico incorporado. En otras situaciones, la apariencia papilar es simplemente un cambio secundario, una mera circunstancia sin ningún tipo de implicaciones en el diagnóstico o en el pronóstico. Conviene, pues, distinguir entre el carcinoma con papilas y el carcinoma papilar, porque ambos términos no son necesariamente sinónimos. Es más, puede haber carcinomas papilares sin papilas, como en el caso del carcinoma papilar de tiroides.
Esta revisión contempla el cáncer renal desde el mero aspecto morfológico papilar. Hay que resaltar que muchos órganos pueden desarrollar carcinomas y adenomas papilares, aunque la vejiga y el riñón son dos de los más frecuentes. En este trabajo se revisa el espectro de carcinomas renales que pueden mostrar papilas al microscopio, a saber, el carcinoma renal papilar esporádico, el carcinoma renal papilar hereditario, el carcinoma renal asociado a leiomiomatosis hereditaria, el carcinoma renal papilar asociado a carcinoma papilar tiroideo hereditario, y algunos carcinomas asociados, bien a la enfermedad de Birt-Hogg-Dubé, o a translocaciones Xp11.2.
CARCINOMA RENAL PAPILAR ESPORÁDICO
Aproximadamente el 10 a 15% de los carcinomas renales primarios son carcinomas papilares (3,4). En comparación con el carcinoma renal de células claras, el carcinoma renal papilar tiene mejor comportamiento biológico (5). La primera descripción detallada de estos tumores apareció en 1976 (6). Con posterioridad, la clasificación morfológica de los tumores renales propuesta por Thoenes y cols (7) los denominó carcinomas renales cromófilos debido a la basofilia citoplásmica que exhibían. La clasificación de Mainz (8) supuso una evolución integrando los primeros hallazgos citogenéticos.
El primer intento de separar los carcinomas renales papilares en dos subgrupos en función de datos exclusivamente morfológicos corrió a cargo de Delahunt y Eble (9). Muy poco después dicha subclasificación histológica se reforzó con el hallazgo de alteraciones genéticas específicas de cada uno de los tipos (10).
Macroscopía
Se trata de neoplasias bien circunscritas y generalmente excéntricas en el riñón. La mayoría están confinadas al riñón y encapsuladas (3). Al corte son tumores de coloración heterogénea dependiendo del grado de necrosis, hemorragia, y de la cantidad de histiocitos espumosos acumulada, variando desde el color amarillento hasta en marrón o rojizo. Es típico encontrar necrosis y hemorragia extensas, aunque este hecho no parece conferir peor pronóstico al carcinoma papilar (11). La necrosis puede ser tan extensa que la imagen radiológica puede ser enteramente quística y los radiólogos a veces encuentran dificultades para etiquetar la masa como un tumor. El examen macroscópico en estos casos con necrosis extensa obliga a una minuciosa búsqueda de zonas conservadas, generalmente en la periferia, para encontrar tejido viable y poder diagnosticas mejor. Los carcinomas papilares se asocian con mayor frecuencia que los demás tumores renales a adenomas corticales. La multifocalidad, encontrada con cierta frecuencia en este tipo de tumor no parece conferir peor pronóstico, y su presencia no debe ser un argumento contra la cirugía conservadora (12). Existen casos aislados que asocian carcinomas y adenomas papilares múltiples en los que algunos tumores muestran histologías mixtas, a caballo entre adenoma y carcinoma, sugiriendo una transición entre ambos (13).
Histología
El carcinoma renal papilar tipo 1 es el más frecuente (fig. 1). Está constituido por células pequeñas, con escaso citoplasma basófilo y núcleo redondeado con nucleolo poco aparente, que se aponen en hilera simple en el eje hialino-vascular de la papila (fig. 2). Se trata de tumores de grado bajo que contienen abundantes acúmulos de histiocitos espumosos (fig. 3) y cuerpos de psammoma en las papilas.
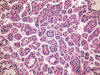
Fig. 1:
Crecimiento papilar típico en el carcinoma renal
papilar esporádico de tipo 1.
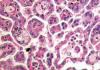
Fig. 2:
Detalle histológico de las células basofílicas de morfología cuboidea del
carcinoma renal papilar esporádico de tipo 1.
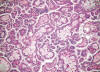
Fig. 3:
Acúmulo de histiocitos espumosos en el carcinoma renal papilar esporádico de
tipo 1.
El carcinoma renal papilar tipo 2 se caracteriza por papilas irregulares (fig. 4) y células de gran tamaño con citoplasma amplio y eosinofílico y núcleo voluminoso con nucleolo prominente (fig. 5). Las células a menudo muestran imágenes de pseudoestratificación en las papilas. Los cuerpos de psammoma (fig. 6) y los acúmulos histiocitarios aparecen en menor cantidad que en el tipo 1.
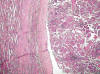
Fig. 4:
Crecimiento papilar típico en el carcinoma renal papilar esporádico de tipo 2.
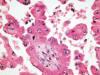
Fig. 5:
Detalle histológico de las células eosinofílicas de morfología columnar alta,
con atipia, del carcinoma renal papilar esporádico de tipo 2.
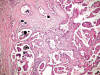
Fig. 6:
Típicos cuerpos de psammoma en el carcinoma renal papilar esporádico de tipo 2.
Normalmente el grado (14) es mayor en el tipo 2 que en el tipo 1 (15). Recientemente se ha propuesto que los carcinomas renales papilares deben ser graduados en base al grado nucleolar y no mediante el sistema de Furhman (16). Asimismo, el estadio TNM (17) en más avanzado en el tipo 2 (15). Como el resto de carcinomas renales, el carcinoma papilar puede presentar transformación sarcomatoide (18). En ocasiones es de morfología sólida (19) o mixta, tubular y papilar y, más raramente, de predominio tubular y en este terreno pueden producirse ciertas dificultades diagnósticas. A veces muestran transformación oncocítica de sus células (20) (fig. 7).
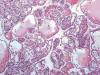
Fig. 7:
Carcinoma renal papilar esporádico mostrando células oncocíticas.
Existen controversias en la literatura reciente sobre si considerar o no al carcinoma fusocelular tubular y mucinoso, un tipo específico de carcinoma renal en la última clasificación de la OMS (4), como un subtipo de carcinoma papilar. Mientras Cossu-Rocca y cols. (21) descartan esta posibilidad basándose en estudios genéticos, Shen y cols. (22) la apoyan.
Inmunohistoquímica
El carcinoma renal papilar comparte con el carcinoma renal de células claras la mayor parte de los marcadores inmunohistoquímicos al uso. Solo la expresión de CK7 (fig. 8) y de p504S (fig. 9) los distingue con fiabilidad, siendo positiva en el primero y negativa en el segundo. Tanto el tipo 1 como el tipo 2 resultan positivos con CD10, marcador de carcinoma renal, citoqueratinas de bajo peso y p504S, y negativos para CK20 y citoqueratinas de alto peso molecular. El tipo 1, además, es positivo con CK7 y MUC1 (23), y el tipo 2 lo es con e-caderina (24).
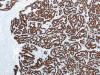
Fig. 8:
Intensa inmunoreactividad con CK 7 en el carcinoma renal papilar tipo 1.

Fig. 9:
Inmunoreactividad con p504S en el carcinoma renal papilar de tipo 1.
Genética
Tradicionalmente, la histología ha sido el método diagnóstico de elección en los carcinomas renales y en las últimas décadas la inmunohistoquímica ha venido a reforzar este punto de vista morfológico. Sin embargo, aunque la combinación de ambos ofrece buenos resultados en la práctica muestran ciertas limitaciones en algunas situaciones. Éstas vienen dadas por al menos dos hechos (25); el primero es que existe un solapamiento morfológico muy significativo entre los diversos tipos reconocidos, y el segundo es que la evaluación histológica es subjetiva y depende demasiado de la experiencia del patólogo evaluador. Es en este contexto en donde el perfil molecular de cada neoplasia tiene un valor innegable (25).
El carcinoma renal papilar está caracterizado por presentar trisomías o tetrasomías de los cromosomas 7 y 17 y por pérdida del cromosoma Y (8,26). Asimismo, se detectan ganancias cromosómicas de 3q, 12, 16 y 20 (26,27). La trisomía 7 es característica pero no específica y se puede encontrar también en tumores de vejiga, próstata y de la cabeza y cuello, en la hiperplasia adenomiomatosa de próstata y en otras enfermedades benignas. Sin embargo es una alteración muy constante tanto en las fases tempranas como tardías de la enfermedad, lo que sugiere que los genes situados en el cromosoma 7 juegan un papel fundamental en la patogenia de estos tumores (28). La trisomía 17 es mucho más específica y valorable desde el punto de vista diagnóstico, pudiendo encontrarse trisomías completas, isocromosomas 17q, o duplicaciones en 17q21.
El análisis genético del carcinoma renal papilar sirve además como elemento predictivo de pronóstico y para ahondar en el conocimiento de la neoplasia. Así, los carcinomas papilares con pérdida de Xp (10) o de 9p (29) tienen mal pronóstico, con supervivencias acortadas. Además, Gunawan y cols (29) demuestran mediante estudios citogenéticas que el tipo 2 de carcinoma renal papilar surge de una evolución a partir del tipo 1, y Jones y cols. (30) encuentran que los carcinomas papilares multifocales tienen un origen independiente, a diferencia de los carcinomas de células claras multifocales, que se consideran de origen único y en los que la multifocalidad se explicaría como un proceso metastático intrarenal.
CARCINOMA RENAL PAPILAR HEREDITARIO
El carcinoma renal papilar hereditario es una enfermedad autosómica dominante que se caracteriza por tumores bilaterales múltiples de comienzo tardío, aunque existen casos aislados de aparición temprana (31). La enfermedad muestra un curso indolente. Muchos pacientes están asintomáticos al diagnóstico, y son descubiertos en exámenes radiológicos rutinarios o realizados por otras causas. Radiológicamente son tumores hipovasculares y su imagen varía poco tras la administración de contraste (32).
Histología
La enfermedad se caracteriza por múltiples tumores de morfología papilar o túbulo-papilar. Se observa toda la gama, desde pequeños grupos microscópicos y adenomas papilares, hasta carcinomas papilares de tipo 1 (33). Las células malignas muestran bajo grado nuclear. El patrón histológico tubular y papilar puede presentar áreas sólidas semejando al adenoma metanéfrico. Los ejes papilares son finos y contienen, al igual que los carcinomas papilares esporádicos, acúmulos de histiocitos espumosos y cuerpos de psammoma.
Curiosamente, hasta el 70% de los carcinomas papilares hereditarios contienen células claras (33) (fig. 10), en especial en aquellas áreas con necrosis o hemorragia. Las células claras muestran un citoplasma a veces espumoso y un núcleo cromático, con nucleolo basófilo. El carácter eminentemente papilar de la neoplasia, junto a la ausencia del patrón vascular típico, ayuda a distinguir el carcinoma papilar renal hereditario con células claras del carcinoma renal de células claras. El resto del diagnóstico diferencial incluye al resto de carcinomas papilares, al carcinoma de los túbulos colectores con predominio papilar y, aquellos casos con un crecimiento preferentemente sólido, con el adenoma matanéfrico. Este último carece de las alteraciones genéticas propias del carcinoma papilar (34,35).
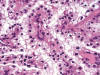
Fig. 10:
Células claras y arquitectura papilar en el carcinoma renal papilar hereditario.
Genética
El carcinoma renal papilar hereditario está causado por mutaciones en el proto-oncogén c-MET, que está constituido por 20 exones y se localiza en 7q31-34 (36,37). Codifica una tirosin-kinasa que está implicada en diversas respuestas biológicas relacionadas con el control del crecimiento celular, la supervivencia celular y la morfogénesis (38). c-MET y su ligando, el factor de crecimiento hepatocitario, son necesarios para el desarrollo embrionario normal del músculo y del hígado y su ausencia es incompatible con la vida (39).
La sobre-expresión de c-MET, al igual que la de sus proto-oncogenes análogos c-kit y RET, está asociado a múltiples neoplasias (40). Las alteraciones responsables del carcinoma renal papilar hereditario suelen ser bien mutaciones en línea germinal a nivel de los exones 16 a 19, o bien trisomías 7. Un pequeño subgrupo de carcinomas renales papilares esporádicos también presenta mutaciones de c-MET idénticas a las de las formas hereditarias (33,34).
CARCINOMA RENAL ASOCIADO A LEIOMIOMATOSIS HEREDITARIA
El síndrome de leiomiomatosis hereditaria y carcinoma renal asocia leiomiomas cutáneos y uterinos junto con carcinoma renal papilar de tipo 2, y fue descrito en 2001, cuando se describieron tres familias susceptibles a desarrollar leiomiomas cutáneos y uterinos (41-44). Aproximadamente el 30% de estos pacientes desarrollan un único carcinoma renal de alto grado de malignidad, al contrario que ocurre en el resto de carcinomas renales asociados a síndromes heredados. Estos carcinomas presentan alta agresividad biológica, con alta frecuencia de metástasis en el momento del diagnóstico. Histológicamente son superponibles al tipo 2 de carcinoma papilar esporádico, aunque se han publicados casos de carcinoma de los ductos colectores y oncocitomas (45).
Histología
El carcinoma renal desarrollado en este síndrome es del tipo papilar o túbulo-papilar (fig. 11). Las células muestran alto grado nuclear, con pleomorfismo marcado. De forma típica, estas células tienen nucleolos eosinófilos muy prominentes, con halo perinucleolar característico (fig. 12). Los citoplasmas son amplios y eosinófilos y, cuando las células forman túbulos, las luces contienen un material basófilo.
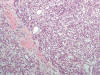
Fig. 11:
Crecimiento tubular en el carcinoma renal asociado a leiomiomatosis hereditaria.
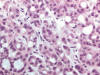
Fig. 12:
Detalle histológico del carcinoma renal papilar asociado a leiomiomatosis
hereditaria mostrando nucleolos eosinofílicos prominentes y halo perinuclear.
Genética
El síndrome de leiomiomatosis hereditaria y carcinoma renal es autosómico dominante. El gen afectado contiene 10 exones, está localizado en 1q42.3-43 y codifica la fumarato hidratasa, una enzima del ciclo de Krebs que convierte el fumarato en malato (43). Se trata de un gen supresor tumoral y la enfermedad se desarrolla en estos pacientes cuando el segundo alelo sufre una mutación somática (42). Aún no se ha descubierto la relación exacta existente entre el déficit de fumarato hidratasa y la génesis de este tipo de carcinoma renal tan característico, aunque la acumulación de glutation intracelular como consecuencia del defecto en la enzima podría estar detrás del inicio del proceso neoplásico (46).
CARCINOMA RENAL PAPILAR ASOCIADO A CARCINOMA PAPILAR DE TIROIDES HEREDITARIO
Recientemente se ha descrito una familia con carcinoma papilar de tiroides hereditario en la cual dos miembros desarrollaron carcinoma papilar renal con mutación en c-MET similar a la del carcinoma papilar renal hereditario comentado en párrafos anteriores (47). La posibilidad de encontrar alteraciones del c-MET en cáncer tiroideo ya ha sido comentada ocasionalmente en la literatura (48,49) sugiriendo mecanismos etiopatogénicos comunes. Asimismo, se ha relacionado con formas agresivas (48) o con elevada tasa de recidiva del carcinoma de tiroides (50).
Sin embargo, la alteración genética que conexiona los carcinomas papilares de ambos órganos parece estar localizada en 1q21 (47), región cromosómica en la que se localizan varios oncogenes, entre ellos NRAS, PRCC (oncogén implicado en el carcinoma renal papilar) y NTRK1 (tirosin quinasa implicada en el carcinoma papilar de tiroides) (51).
CARCINOMA PAPILAR ASOCIADO AL SÍNDROME DE BIRT-HOGG-DUBÉ
El síndrome de Birt-Hogg-Dubé es una enfermedad autonómica dominante que consiste en una triada de lesiones cutáneas compuesta por fibrofoliculomas, tricodiscomas, y acrocordones (52), al que se le añaden con cierta frecuencia neumotórax espontáneos, neoplasias colorectales y diversos tipos de carcinomas renales entre los que predominan los carcinomas de células cromófobas (53). Muy raramente se han descrito carcinomas papilares en este síndrome y corresponden histológicamente al tipo 2 (54).
La alteración genética en este síndrome se localiza en el cromosoma 17 (17p12-q12.2) (55).
CARCINOMAS CON FUSIÓN ASPL-TFE3
Con este nombre se agrupan una serie de carcinomas renales que se presentan en la edad pediátrica y que tienen en común una translocación en la que el punto de ruptura está en Xp11.2. Muchos de estos pacientes se caracterizan por haber recibido, varios años antes, tratamientos de quimioterapia citotóxica por leucemias y otros tumores, e incluso por lupus eritematoso sistémico (56).
El carcinoma renal con fusión ASPL-TFE3 [t(X,17)(p11.2;q25)] es el más típicamente papilar entre todos los carcinomas con translocación Xp11.2. Se presenta en estadios avanzados y en pacientes por debajo de los 18 años de edad. Casi todos los casos descritos muestran metástasis ganglionares a pesar del pequeño tamaño del tumor primario. Sin embargo, muestran un curso clínico indolente.
Histología
Los carcinomas renales con translocación Xp11.2 tienen una morfología típica. Se trata de carcinomas compuestos por células claras de gran tamaño que forman papilas (fig. 13), nidos y alvéolos acompañados de abundantes cuerpos de psammoma. A veces presentan citoplasmas levemente eosinófilos. Todos estos carcinomas, antes de ser evaluados e identificados genéticamente, pertenecían al raro grupo de carcinomas renales de la infancia y adolescencia. De forma característica, estos tumores son positivos con los marcadores inmunohistoquímicos del melanoma (Melan A y HMB45) y no expresan, o lo hacen muy débilmente, citoqueratinas y vimentina (56,57). También se detecta positividad con CD10 y con el marcador del carcinoma renal. Más específicamente, este grupo de carcinomas muestra positividad nuclear con el factor de transcripción TFE3 en inmunohistoquímica de material incluido en parafina. Al microscopio electrónico estos carcinomas contienen gránulos citoplásmicos rodeados por membrana y cristales romboideos similares a los que se detectan en el sarcoma alveolar de partes blandas (58).
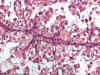
Fig. 13:
Células claras irregulares con núcleos atípicos y papilas en el carcinoma renal
con translocación Xp11.2.
Genética
Como consecuencia de la rotura en Xp11.2 se producen fusiones del factor de transcripción TFE3, que es el que se codifica precisamente en esa región cromosómica, con otros genes (57,58). Hasta la fecha se han descrito 5 diferentes fusiones que afectan al TFE3 (58,59). Entre ellas, la ASPL-TFE3 es idéntica a la que se produce en el sarcoma alveolar de partes blandas (58).
CONCLUSIONES
La clasificación de los tumores renales de la OMS publicada en 2004 reconoce 49 tipos diferentes de neoplasias renales (60). Hasta la fecha, las clasificaciones de los tumores renales se basaban fundamentalmente en parámetros histológicos. Sin embargo, el espectro de carcinomas renales que muestran morfología papilar incluye un grupo heterogéneo de tumores asociados a muy diversas alteraciones genéticas. Este trabajo revisa todo este espectro.
BIBLIOGRAFÍA
Fletcher CDM. Diagnostic Histopathology of Tumors. 3rd Edition. London: Churchill Livingstone; 2007.
Rosai J. Rosai and Ackerman’s Surgical Pathology. 9th Edition. Edinburgh: Mosby; 2004.
Murphy WM, Grignon DJ, Perlman EJ. Tumors of the Kidney, Bladder, and Related Urinary Structures. AFIP Atlas of Tumor Pathology, 4th Series, Fascicle 1. Washington DC: American Registry of Pathology; 2004.
López-Beltrán A, Scarpelli M, Montironi R, Kirkali Z. 2004 WHO classification of the renal tumors of the adults. Eur Urol 2006; 49: 798-805.
Pignot G, Elie C, Conquy S, Vieillefond A, Flam T, Zerbib M, et al. Survival analysis of 130 patients with papillary renal cell carcinoma: Prognostic utility of type 1 and type 2 subclassification. Urology 2007; 69: 230-5.
Mancilla-Jimenez R, Stanley RJ, Blath RA. Papillary renal cell carcinoma. A clinical, radiologic and pathologic study of 34 cases. Cancer 1976; 38: 2469-80.
Thoenes W, Störkel S, Rumpelt HJ. Histopathology and classification of renal cell tumors (adenomas, oncocytomas and carcinomas): The basic cytological and histopathological elements and their use in diagnostics. Pathol Res Pract 1986; 181: 125-43.
Kovacs G. Molecular differential pathology of renal cell tumours. Histopathology 1993; 22: 1-8.
Delahunt B, Eble JN. Papillary renal cell carcinoma: A clinicopathologic and immunohistochemical study of 105 tumors. Mod Pathol 1997; 10: 537-544.
Jiang F, Richter J, Schraml P, Bubendorf L, Gasser T, Sauter G, et al. Chromosomal imbalances in papillary renal cell carcinoma: Genetic differences between histologic types. Am J Pathol 1998; 153: 1467-73.
Lee SE, Byun SS, Oh JK, Lee SC, Chang IH, Choe G, et al. Significance of macroscopic tumor necrosis as a prognostic indicator for renal cell carcinoma. J Urol 2006; 176: 1332-7.
Mejean A, Hopirtean V, Bazin JP, Larousserie F, Benoit H, Chrétien Y, et al. Prognostic factors for the survival of patients with papillary renal cell carcinoma: meaning of histological typing and multifocality. J Urol 2003; 170: 764-7.
Kiyoshima K, Oda Y, Nakamura T, Migita T, Okumura K, Naito S, et al. Multicentric papillary renal cell carcinoma associated with renal adenomatosis. Pathol Int 2004; 54: 266-72.
Furhman SA, Lasky LC, Limas C. Prognostic significance of morphologic parameters in renal cell carcinoma Am J Surg Pathol 1982; 6: 655-63.
Delahunt B, Eble JN, McCredie MR, Bethwaite PB, Stewart JH, Bilous AM. Morphologic typing of papillary renal cell carcinoma: Comparison of growth kinetics and patient survival in 66 cases. Hum Pathol 2001; 32: 590-5.
Sika-Paotonu D, Bethwaite PB, McCredie MR, William Jordan T, Delahunt B. Nucleolar grade but not Furhman grade is applicable to papillary renal cell carcinoma. Am J Surg Pathol 2006; 30: 1091-6.
Sobin LH, Wittekind C. TNM classification of Malignant Tumours, 6th edition. New Jersey: John Wiley & Sons; 2002.
Cohen RJ, McNeal JE, Susman M, Sellner LN, Iacopetta BJ, Weinstein SL, et al. Sarcomatoid renal cell carcinoma of papillary origin. A case report and cytogenetic evaluation. Arch Pathol Lab Med 2000; 124: 1830-2.
Renshaw AA, Zhang H, Corless CL, Fletcher JA, Pins MR. Solid variants of papillary (chromophil) renal cell carcinoma: clinicopathologic and genetic features. Am J Surg Pathol 1997; 21: 1203-9.
Lefèvre M, Couturier J, Sibony M, Bazille C, Boyer K, Callard P, et al. Adult papillary renal tumor with oncocytic cells. Clinicopathologic, immunohistochemical, and cytogenetic features of 10 cases. Am J Surg Pathol 2005; 29: 1576-81.
Cossu-Rocca P, Eble JN, Delahunt B, Zhang S, Martignoni G, Brunelli M, et al. Renal mucinous tubular and spindle carcinoma lacks the gains of chromosomes 7 and 17 and losses of chromosome Y that are prevalent in papillary renal cell carcinoma. Mod Pathol 2006; 19: 488-93.
Shen SS, Ro JY, Tamboli P, Truong LD, Zhai Q, Jung SJ, Tibbs RG, et al. Mucinous tubular and spindle cell carcinoma of kidney is probable a variant of papillary renal cell carcinoma with spindle cell features. Ann Diagn Pathol 2007; 11: 13-21.
Leroy X, Zini L, Leteurtre E, Zerimech F, Porchet N, Aubert JP, et al. Morphologic subtyping of papillary renal cell carcinoma: correlation with prognosis and differential expresión of MUC1 between the two subtypes. Mod Pathol 2003; 15: 1126-30.
Zhou M, Roma A, Magi-Galluzzi C. The usefulness of immunohistochemical markers in the differential diagnosis of renal neoplasms. Clin Lab Med 2005; 25: 247-57.
Yang XJ, Sugimura J, Schafernak KT, Tretiakova MS, Han M, Vogelzang NJ, et al. Classification of renal neoplasms based on moleculalr signatures. J Urol 2006; 175: 2302-6.
Kovacs G, Füzesi L, Emanual A, Kung HF. Cytogenetics of papillary renal cell tumors. Genes Chromosomes Cancer 1991; 3: 249-55.
Henke RP, Erbersdobler A. Numerical chromosomal aberrations in papillary renal cortical tumors. Relationship with histopathologic features. Virchows Arch 2002; 440: 604-9.
Glukhova L, Lavialle C, Fauvet D, Chudoba I, Danglot G, Angevin E, et al. Mapping of the 7q31 subregion common to the small chromosome 7 derivatives from two sporadic papillary renal cell carcinomas: increased copy number and overexpression of the MET proto-oncogene. Oncogene 2000; 19: 754-61.
Gunawan B, von Heydebreck A, Fritsch T, Huber W, Ringert RH, Jakse G, et al. Cytogenetic and morphologic typing of 58 papillary renal cell carcinomas: evidence for a cytogenetic evolution of Type 2 from Type 1 tumors. Cancer Res 2003; 63: 6200-5.
Jones TD, Eble JN, Wang M, MacLennan GT, Delahunt B, Brunelli M, et al. Molecular genetic evidence for the independent origin of multifocal papillary tumors in patients with papillary renal cell carcinomas. Clin Cancer Res 2005; 11: 7226-33.
Schmidt LS, Nickerson ML, Angeloni D, Glenn GM, Walther MM, Albert PS, et al. Early onset hereditary papillary renal carcinoma: germline missense mutations in the tyrosine kinase domain of the met proto-oncogene. J Urol 2004; 172: 1256-61.
Choyke PL, Walther MM, Glenn GM, Wagner JR, Venzon DJ, Lubensky IA, et al. Imaging features of hereditary papillary renal cancers. J Comput Assist Tomogr 1997; 21: 737-41.
Lubensky IA, Schmidt L, Zhuang Z. Hereditary and sporadic papillary renal cell carcinomas with c-met mutations share a distinct morphological phenotype. Am J Pathol 1999; 155: 517-26.
Schmidt L, Junker K, Nakaigawa N, Kinjerski T, Weirich G, Miller M, et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 1999; 18: 2343-50.
Brunelli M, Eble JN, Zhang S, Martignoni G, Cheng L. Metanephric adenoma lacks the gains of chromosomes 7 and 17 and loss of Y that are typical of papillary renal cell carcinoma and papillary adenoma. Mod Pathol 2003; 16: 1060-3.
Schmidt L, Duh FM, Chen F, Kishida T, Glenn G, Choyke P, et al. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat Genet 1997; 16: 68-73.
Bottaro DP, Linehan WM. Multifocal renal cancer: genetic basis and its medical relevance. Clin Cancer Res 2005; 11: 7206-8.
Cohen D, Zhou M. Molecular genetics of familial renal cell carcinomas syndromes. Clin Lab Med 2005; 25: 259-77.
Ponzetto C, Pante G, Prunotto' C, Ieraci A, Maina F. Met signaling mutants as tools for developmental studies. Int J Dev Biol 2000; 44: 645-53.
Di Renzo MF, Narsimhan RP, Olivero M, Bretti S, Giordano S, Medico E, et al. Expression of the Met/HGF receptor in normal and neoplastic human tisúes. Oncogene 1991; 6: 1997-2003.
Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, Sistonen P, Herva R, Aaltonen LA. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci USA 2001; 98: 3387-92.
Kiuru M, Launonen V, Hietala M, Aittomäki K, Vierimaa O, Salovaara R, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol 2001; 159: 825-9.
Alam NA, Rowan AJ, Wortham NC, Pollard PJ, Mitchell M, Tyrer JP,et al. Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, hereditary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 2003; 12:1241-52.
Linehan WM, Vasselli J, Srinivasan R, Walther MM, Merino M, Choyke P, et al. Genetic basis of cancer of the kidney: disease-specific approaches to therapy. Clin Cancer Res 2004; 10: 6282-9.
Toro JR, Nickerson ML, Wei MH, Warren MB, Glenn GM, Turner ML, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America. Am J Hum Genet 2003; 73: 95-106.
Matés JM, Pérez-Gómez C, Núñez de Castro I, Asenjo M, Márquez J. Glutamine and its relationship with intracellular redox status, oxidative stress and cell proliferation/death. Int J Biochem Cell Biol 2002; 34: 439-58.
Malchoff CD, Sarfarazi M, Tendler B, Forouhar F, Whalen G, Joshi V, et al. Papillary thyroid carcinoma associated with papillary renal neoplasia: genetic linkage analysis of a distinct heritable tumor syndrome. J Clin Endocrinol Metab 2000; 85: 1758-64.
Nardone HC, Ziober AF, Livolsi VA, Mandel SJ, Baloch ZW, Weber RS, et al. c-Met expression in tall cell variant papillary carcinoma of the thyroid. Cancer 2003; 98: 1386-93.
Wasenius VM, Hemmer S, Karjalainen-Lindsberg ML, Nupponen NN, Franssila K, Joensuu H. MET receptor tyrosine kinase sequence alterations in differentiated thyroid carcinoma. Am J Surg Pathol 2005 29: 544-9.
Inaba M, Sato H, Abe Y, Umemura S, Ito K, Sakai H. Expression and significance of c-met protein in papillary thyroid carcinoma. Tokai J Exp Clin Med 2002; 27: 43-9.
Moore FD Jr. Inherited aspects of papillary thyroid carcinoma. J Surg Oncol 2006; 94: 719-24.
Birt A, Hogg GR, Dubé WJ. Hereditary multiple fibrofolliculomas with trichodiscomas and acrochordons. Arch Ddermatol 1977; 113: 1674-7.
Khoo SK, Kahnoski K, Sugimura J, Petillo D, Chen J, Shockley K, et al. Inactivation of BHD in sparadic renal tumors. Cancer Res 2003; 63: 4583-7.
Pavlovich CP, Walther MM; Eyler RA, Hewitt SM, Zbar B, Linehan WM, et al. Renal tumors in the Birt-Hogg-Dubé syndrome. Am J Surg Pathol 2002; 26: 1542-52.
Khoo SK, Bradley M, Wong FK, Hedblad MA, Nordenskjöld M, Teh BT. Birt-Hogg-Dubé syndrome: mapping of a novel hereditary neoplasia gene to chromosome 17p12-q11.2. Oncogene 2001; 20: 5239-42.
Argani P, Lae M, Ballard ET, Amin M, Manivel C, Hutchinson B, et al. Translocation carcinomas of the kidney after chemotherapy in childwood. J Clin Oncol 2006; 24: 1529-34.
Argani P, Laé M, Hutchinson B, Reuter VE, Collins MH, Perentesis J, et al. Renal carcinomas with the t(6;11)(p21;q12). Clinicopathologic features and demonstration of the specific alpha-TFEB gene fusion by immunohistochemistry, RT-PCR, and DNA PCR. Am J Surg Pathol 2005; 29: 230-40.
Argani P, Hawkins A, Griffin CA, Goldstein JD, Haas M, Beckwith JB, et al. A distinctive pediatric renal neoplasm characterized by epithelioid morphology, basement membrane production, focal HMB45 immunoreactivity, and t(6;11)(p21.1;q12) chromosome translocation. Am J Pathol 2001; 158: 2089-96.
Argani P, Antonescu CR, Couturier J, Fournet JC, Sciot R, Debiec-Rychter M, et al. PRCC-TFE3 renal cell carcinomas: morphologic, immunohistochemical, ultrastructural and molecular analysis o fan entity associated with the t(X;1)(p11.2;q21). Am J Surg Pathol 2002, 26: 1553-66.
Eble JN, Sauter G, Epstein JI, Sesterhenn IA. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs. Lyon: IARC Press; 2004.
![]()