
Vol. 39, n.º 3, 2006
|
REVISTA
ESPAÑOLA DE
Vol. 39, n.º 3, 2006 |
REVISIONES
Juan Segura Sánchez1, María Jesús Pareja Megía2, Antonio García Escudero2, María Teresa Vargas de los Monteros2, Ricardo González-Cámpora2
1 Servicio de Anatomía Patológica.
Hospital Infanta Margarita. Cabra, Córdoba.
2 Departamento de Anatomía Patológica y Citogenética. Hospital
Universitario Virgen Macarena. Sevilla.
rcampora@us.es
RESUMEN
Los liposarcomas constituyen el grupo más numeroso de sarcomas del adulto. Su interés actual radica fundamentalmente en los cambios conceptuales y clasificatorios que han acontecido en los últimos años merced a la aplicación de las técnicas de citogenética y de biología molecular. En la presente revisión se lleva a cabo una correlación clínico patológica de los cinco tipos básicos de liposarcomas y se comentan los aspectos citogenéticos y de biología molecular que han permitido la elaboración de la nueva clasificación de la OMS.
Palabras claves: Liposarcomas, citogenética, biología molecular.
SUMMARY
Liposarcomas are the most common sarcoma of the adult life. Their current interest is based on the recent molecular and cytogenetic changes that have allowed the new WHO classification. In the present report we carried out a clinicopathological correlation in the five distinctive groups of the current classification and described the most relevant cytogenetics and molecular findings in each group.
Key words: Liposarcoma, cytogenetics, molecular biology.
Los tumores del tejido adiposo constituyen uno de los grupos más numerosos de neoplasias de los tejidos blandos y los liposarcomas, en particular, son los sarcomas más comunes en el adulto (1). El interés actual por estas neoplasias no radica en su elevada incidencia sino en que han sido objeto de importantes cambios conceptuales y clasificatorios en los últimos años, merced a la aplicación de las técnicas de citogenética y de biología molecular (2,3). Es de destacar que, en contraposición con los otros tumores de los tejidos blandos, el diagnóstico descansa principalmente en la correlación clínica y en el uso de las técnicas histológicas clásicas, siendo muy escasa la contribución aportada por la inmunohistoquímica.
En la tabla 1 aparecen recogidas de modo comparativo las clasificaciones de los tumores lipomatosos de la OMS de las dos últimas ediciones, 1994 y 2002 (3,4). Los cambios más significativos los podemos resumir en los siguientes puntos:

— Tumores benignos:
Aparición de dos nuevas formas peculiares de lipomas: el lipoma condroide y el miolipoma.
Cambio en la denominación del fibrolipoma del nervio por el de lipomatosis del nervio.
— Tumores malignos: liposarcomas (LPS):
Clasificación en 4 subtipos, incluidas las formas mixtas e inclasificables.
Inclusión de un nuevo subtipo de LPS: el liposarcoma desdiferenciado (LPS DD), que se considera la evolución maligna del Liposarcoma bien diferenciado (LPS BD)/tumor lipomatoso atípico (TLA).
Agrupación de los liposarcomas mixoides (LPS MIX) y de células redondas (LPS CR) en una única entidad definida por las alteraciones moleculares.
— Creación de una nueva categoría biológica: Tumor de malignidad intermedia para aquellas lesiones potencialmente malignas que carecen de capacidad metastatizante: tumor lipomatoso atípico/liposarcoma bien diferenciado (TLA/LPS BD).
LIPOSARCOMAS
En conjunto, los LPS representan casi el 20% de todos los sarcomas del adulto (1,2,5). Aunque los distintos subtipos reconocidos por la OMS son entidades totalmente independientes, que sólo tienen en común su histogénesis, se pueden realizar las siguientes generalizaciones:
Al contrario que los lipomas, mayoritariamente son lesiones profundas por lo que es poco probable que un lipoma pueda degenerar en LPS. No obstante, algunos autores (6) han teorizado sobre la posibilidad de una progresión tumoral: lipoma, TLA/LPS BD, LPS DD.
Son tumores propios de la edad adulta (incidencia máxima entre la 5.ª-7.ª décadas) siendo excepcionales en niños. Los casos descritos en la infancia se han presentado en niños entre los 10-15 años, son de tipo mixoide y han cursado siempre con pronóstico favorable (7).
Las técnicas de IHQ e histológicas especiales tienen escaso valor en el diagnóstico cotidiano; sólo la proteína S-100 puede ser de cierta ayuda en el reconocimiento de las formas más complicadas de LPS MIX (variedad de células redondas).
Aunque durante muchos años el reconocimiento del lipoblasto ha sido clave para el diagnóstico de LPS, en la actualidad su importancia se ha reducido notablemente ya que no siempre están presentes y también pueden encontrarse, incluso en número considerable, en ciertas lesiones benignas: lipoblastoma, lipoma pleomórfico, lipoma condroide y lipoma de células fusiformes; además, hay lesiones no lipomatosas (tumorales o reactivas) que contienen células con morfología totalmente superponibles a los lipoblastos con las técnicas rutinarias. Por tanto, el valor diagnóstico del lipoblasto sólo se hace patente en un contexto adecuado (1,8).
El lipoblasto es una célula mesenquimal poco desarrollada que muestra diferenciación adiposa; morfológicamente se reconoce por la presencia de una o varias vacuolas lipídicas que indentan o festonean el contorno nuclear, a la vez que causan discreta hipercromasía y desplazan el núcleo a la periferia (fig. 1A). La tendencia a la maduración se refleja en la fusión de las vacuolas. La célula madura (lipocito) presenta una única vacuola que comprime y desplaza totalmente el núcleo a la periferia (fig. 1B). Los lipocitos o lipoblastos univacuolados no deben confundirse con pequeños vasos capilares de luces abiertas ni con células endoteliales epitelioides vacuolizadas. De igual modo, tampoco deben identificarse como lipoblastos a macrófagos multivacuolados con vacuolas pequeñas que ocupan todo el citoplasma y no modifican el contorno nuclear (fig. 1C). Las células que más remedan a lipoblastos son los macrófagos de los granulomas de silicona; estas células pueden ser bi o multivacuoladas y muestran núcleos hipercromáticos y deformados (fig. 1D); las técnicas de inmunohistoquímica son de gran ayuda en su reconocimiento, puesto que la inmunoreacción a la proteína S-100 es positiva en los lipoblastos y negativa en los macrófagos y, viceversa, la reacción al antígeno CD68 es positiva en las células macrofágicas y negativa en los lipoblastos.
![]()
Fig. 1:
Lipoblastos, lipocitos, macrófagos espumosos y pseudolipoblastos. A. Lipoblastos
multivacuolados con deformidad del contorno nuclear. HE 60x. B. Células
univacuoladas con núcleo desplazado a la periferia (lipocitos) y ocasionales
lipoblastos HE 60x. C. Macrófago multivacuolado de aspecto espumoso. HE 60x. D.
Macrófagos tipo pseudolipoblastos propios del granuloma de silicona. HE 60x.
1. Tumor lipomatoso atípico/Liposarcoma bien diferenciado (TLA/LPS BD)
Se define como un tumor de malignidad intermedia compuesto, totalmente o en parte, por adipocitos maduros que presentan variaciones significativas en su tamaño y, al menos, atipia nuclear focal, tanto en células estromales como lipoblastos (3). Las células atípicas se reconocen porque destacan sobre las demás y muestran uno o varios núcleos hipercromáticos, grandes y con formas irregulares (fig. 2A); su número es muy variable y pueden identificarse en el seno de los adipocitos, en los septos conectivos, o incluso en las paredes de vasos de mediano calibre (fig. 3); por lo general, el citoplasma es poco relevante pero a veces contiene gotas de triglicéridos que deforman el núcleo (lipoblastos atípicos) (fig. 2B). Por tanto, para realizar el diagnóstico histológico de TLA/LPS BD no es necesaria la presencia de estos lipoblastos atípicos (1,3,8) sino simplemente de células atípicas en su ambiente apropiado.
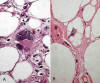
Fig. 2:
A. Célula estromal atípica. HE 60x. B. Lipoblasto
atípico multivacuolado. HE 60x.

Fig. 3:
Células estromales atípicas en la pared de vasos
de mediano calibre. A y B. HE 20x.
Los términos TLA y LPS BD se utilizan indistintamente para denominar a una misma lesión morfológica. El término TLA hace referencia a neoplasias que por su localización superficial o intramuscular (extremidades y tronco) son potencialmente curables mediante cirugía. En cambio, el de LPS BD se utiliza para lesiones profundas (retroperitoneo, mediastino e ingle) donde la cirugía difícilmente es curativa (1,5,9). Aunque existen ciertas reticencias por parte de algunos autores a adoptar esta nomenclatura –los tumores se denominan en base a sus características histológicas y no de acuerdo con el potencial maligno resultante de la localización y de las posibilidades quirúrgicas (8,10)–, en general, hay consenso entre terapeutas y patólogos para no denominar sarcomas a lesiones potencialmente curables, debido a las connotaciones psicológicas y sociales que este término conlleva.
Los TLA/LPS BD representan el 40-50% de todos los LPS (2,3). Se presentan habitualmente en adultos de edad media-avanzada (máxima incidencia en la 6.ª década) y sin diferencias sexuales (1). Por orden de frecuencia, las localizaciones preferenciales son: extremidades (70%) –especialmente el muslo–, retroperitoneo (RP) (20%), área paratesticular y mediastino (10%) (1). Excepcionalmente se han descrito casos en órganos (mama y laringe) (11,12) y tejidos superficiales, incluida la piel (2). En las extremidades, generalmente se manifiestan como masas no dolorosas de crecimiento lento que se hacen particularmente visibles durante la contracción muscular. En el RP suelen pasar desapercibidos hasta llegar a alcanzar grandes dimensiones (>20 cm) o bien causar síntomas compresivos o distensión abdominal. En el cordón espermático se presentan como hernias inguinales (2,8).
Macroscópicamente son lesiones voluminosas, bien circunscritas, de color amarillo-blanquecino en función de la proporción de tejido fibroso existente (1,5,8) (fig. 4). Los márgenes de la lesión suelen ser infiltrativos, de tal modo que se confunden con el tejido graso normal, dificultando su extirpación total; no obstante, tanto en lesiones en RP como intramusculares, no hay evidencias claras de que la «resección incompleta» suponga una mayor tasa de recidivas. En el RP, a veces los tumores se presentan como varios nódulos aparentemente discontinuos.
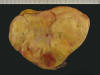
Fig. 4:
Aspecto macroscópico de un liposarcoma bien
diferenciado tipo lipoma con extensas áreas amarillentas de tejido adiposo y una
región central más blanquecina y firme correspondiente tejido fibroso.
Histológicamente se reconocen 4 subtipos específicos (3,5), si bien con frecuencia pueden coexistir dos o más de ellos en una misma lesión, particularmente en el RP. Las formas más comunes son los tipo lipoma y esclerosante que, además, con frecuencia aparecen combinados (1).
El TLA/LPS BD tipo lipoma está compuesto por adipocitos univacuolados relativamente maduros que, en contraste con los lipomas, presentan marcada variabilidad en su tamaño. Además contienen, como hallazgo diagnóstico necesario un número variable de células atípicas (fig. 5). Por lo general, no hay gran dificultad en el reconocimiento de las células atípicas, pero en casos excepcionales pueden llegar a ser muy escasas, de tal modo que se hace necesario un amplio muestreo (1,3,5,8); el diagnóstico se realiza cuando se identifica, al menos, una de ellas. El número de células atípicas con gotas lipídicas (lipoblastos) puede ser muy variable, desde ninguna a muchas.
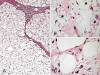
Fig. 5:
Liposarcoma bien diferenciado tipo lipoma. A.
Imagen panorámica donde destaca la variabilidad del tamaño de los adipocitos. HE
2,5x. B. Lipoblastos atípicos. HE 40x. C. Células estromales atípicas. HE 40x.
El TLA/LPS BD tipo esclerosante se observa especialmente en RP y región paratesticular (1,2). Son lesiones hipocelulares con abundante depósito de colágeno, frecuentes células atípicas estromales y ocasionales lipoblastos atípicos. El colágeno de es de tipo fibrilar o cordonal (1-3,5,8) (fig. 6). Puesto que estos territorios pueden llegar a ser muy extensos, a veces es necesario recurrir a nuevos muestreos para identificar focos con adipocitos. No es inusual encontrar áreas mixoides, mixohialinas o incluso diferenciación heteróloga madura, preferentemente ósea u osteocartilaginosa (fig. 7), y más excepcionalmente muscular lisa o estriada (13). Algunos autores (8) admiten la posibilidad de que existan áreas con celularidad moderada e incluso ocasionales figuras de mitosis; no obstante, en la reciente clasificación de la OMS (3) estas lesiones se consideran LPS DD de bajo grado.
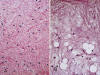
Fig. 6:
Liposarcoma bien diferenciado tipo esclerosante.
A. Detalle de área esclerosante donde destaca el colágeno de tipo cordonal. HE
20x. B Lipoblastos atípicos en el seno de un área esclerosante con colágeno de
tipo fibrilar. HE 40x.

Fig. 7:
Liposarcoma bien diferenciado tipo escelrosante
con metapalasia madura. A. Ósea. HE 20x. B. Cartilaginosa. HE 20x.
El TLA/LPS BD de tipo inflamatorio se observa casi exclusivamente en RP. Viene definido por la coexistencia de marcado infiltrado inflamatorio linfoplasmocitario (fig. 8). Este puede llegar ser tan abundante que impida reconocer el verdadero crecimiento adipocítico y la lesión se confunda con un tumor miofibroblástico inflamatorio, una enfermedad de Castleman o un linfoma de Hodgkin. El infiltrado linfoide es politípico (B y T) y policlonal. La clave diagnóstica, en aquellos casos en que no se identifican fácilmente áreas tipo lipoma o tipo esclerosante, es la presencia de células hipercromáticas atípicas en el seno de la masa tumoral (1-3,5,8,14).
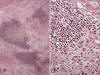
Fig. 8:
Liposarcoma bien diferenciado tipo inflamatorio.
A. Imagen panorámica dodne se advierte un área esclerosante con abundante
infiltrado linfoide. HE 2,5x. Detalle donde se advierte la población
linfoplasmocitaria. HE 20x.
El TLA/LPS BD de tipo células fusiforme es la última forma descrita y la más controvertida. Son lesiones superficiales que asientan preferentemente en el tronco, extremidades y cabeza y cuello. Histológicamente están constituidas por células fusiformes sin atipia, en el seno de un estroma fibroso o mixoide y en asociación con un componente adipocítico, que frecuentemente contiene lipoblastos (3,5,15) (fig. 9). Weiss y Goldblum (1) no lo consideran un subtipo de TLA/LPS sino un patrón histológico propio de LPS MIX. En cualquier caso, no hay que confundirlo con el sarcoma de vainas neurales periférico ni con el lipoma de células fusiformes.

Fig. 9:
Liposarcoma bien diferenciado tipo células
fusiformes. A. Imagen panorámica donde se advierte la proliferación de células
fusiformes con mugrosas células adiposas intercaladas. HE 10x. B. Detalle de las
células fusiformes con adipocitos en su seno. HE 40x.
El diagnóstico diferencial del TLA/LPS BD con el lipoma convencional y con los sarcomas esclerosantes se plantea sólo en situaciones límites. Con respecto al primero punto, el dato fundamental es definir lo que se entiende por atipia en un tumor lipomatoso y cuántas células atípicas deben estar presentes para emitir el diagnóstico de TLA/LPS BD. No hay una contestación definitiva a esta pregunta y, en cierta medida, la respuesta está en función de la localización del tumor. En lesiones no situadas en RP se requiere la presencia de al menos una célula suficientemente grande para destacar de las demás con la lente de 40x. En cambio, en RP, todo tumor lipomatoso compuesto total o parcialmente por células adipocíticas maduras se considera con fines terapéuticos un LPS BD, con independencia de que se identifiquen o no células atípicas. Como norma general, es aconsejable en todos los tumores lipomatosos profundos situados fuera del RP llevar a cabo un amplio muestreo (un bloque por cada cm. de diámetro hasta un máximo de 10) y una búsqueda exhaustiva de células atipicas si la lesión presenta variabilidad en el tamaño de los adipocitos (8).
El diagnóstico diferencial con los sarcomas esclerosantes se presenta en las neoplasias de situación central (RP, mediastino e ingle), donde las lesiones pueden estar constituidas en su totalidad por áreas esclerosantes, mixoides o fibrosas, de tal modo que el componente adipocítico solo se observa focalmente en la periferia o en recidivas posteriores. Evidentemente, sin componente adipocítico no se puede llevar realizar el diagnóstico de LPS, por lo que estas lesiones, en principio, deben considerarse como tumores mixoides o esclerosantes no clasificables y apuntarse la posibilidad de que en futuras recidivas pueda presentarse diferenciación adiposa.
Por último, con respecto a la variedad de LPS BD de células fusiformes, el diagnóstico diferencial con el lipoma de células fusiformes y pleomórficas descansa en las características clínicas (afectación preferencial de los tejidos superficiales del cuello y dorso), histológicas (haces de colágeno cordonal, abundancia de células cebadas) e inmunohistoquímicas (reactividad al CD34) propias de este último (1,3,8).
En la mayoría de las ocasiones las técnicas de inmunohistoquímica no son necesarias para realizar el diagnóstico. No obstante, se ha descrito inmunorreacción positiva a la proteína S-100 en los adipocitos en 2/3 de los casos y, con mucha menos frecuencia, también reactividad al CD34 en las células fusiformes y atípicas.
Los estudios de citogenética, aunque tienen poca relevancia diagnóstica, han sido muy importantes a la hora de considerar esta categoría diagnóstica como entidad clínicopatológica bien definida, ya que existen alteraciones cromosómicas clonales comunes en el 93% de los casos. El hallazgo citogenético más característico es la presencia de anillos y/o megacromosomas extras o dmin (80% de los casos) que contienen secuencias amplificadas de la región cromosómica 12q14-15, precisamente donde se encuentran los genes MDM2, SAS, CDK4 y HMGIC (1-3,8,16,17) (fig. 10). Las alteraciones del cromosoma 12 pueden observarse como única anomalía o bien estar asociadas a otras alteraciones estructurales o numéricas, siendo las más comunes las amplificaciones de los segmentos 12q21-q22 y 1q21-q25 (17). El gen MDM2 aparece frecuentemente amplificado, con o sin co-amplificación de los otros genes, y se puede poner de manifiesto en cortes histológicos parafinados mediante la técnica del FISH o bien a través de su expresión proteica con la técnica de inmunoperoxidasa (3,16-19). Este hallazgo aporta cierto valor diagnóstico en la diferenciación de los TLA/LPS BD tipo lipomas con los lipomas profundos, ya que en estos últimos no existe sobrexpresión proteica de MDM2, auque sí puede haberla de CDK4 y HMGIC (19,20). Con frecuencia, mediante sondas centroméricas del cromosoma 12 también se advierten células aumento del número de copias, indicando amplificaciones de segmentos del cromosoma 12 que incluyen centrómeros (21) (fig. 11A).

Fig. 10:
Metafase de un liposarcoma bien diferenciado tipo
lipoma mostrando el característico cromosoma en anillo.
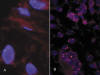
Fig. 11:
Copias centroméricas del cromosoma 12. A.
Liposarcoma bien diferenciado. Adviértase un núcleo con 6 señales, en
contraposición con los restantes donde sólo es patente una o dos. B. Liposarcoma
desdiferenciado. Numerosos núcleos de células tumorales con un gran número de
copias.
Los factores pronósticos más importantes de los TLA/LPSBD son el tiempo de evolución y la localización (1,3,5,8,9). La capacidad de recidiva o desdiferenciación está directamente relacionada con la persistencia clínica de la enfermedad (tiempo). La localización del tumor también es importante, ya que se relaciona con la accesibilidad quirúrgica y la posibilidad de resecciones totales. Así, lesiones periféricas, donde el diagnóstico clínico es más precoz y la accesibilidad quirúrgica mayor, raramente recidivan tras una extirpación con márgenes adecuados y, si lo hacen, son poco agresivas. En los tumores profundos (RP, mediastino, cordón espermático), donde las posibilidades de una resección total son menores, las recidivas son más frecuentes y la muerte puede ser consecuencia de compresión de estructuras vitales o bien por desdiferenciación. EL curso agresivo es particularmente notable en las lesiones situadas en RP donde las recidivas y la desdiferenciación alcanzan el 91% y 17% respectivamente, en contraposición con lo observado en las extremidades donde no superan el 43% y 6%, respectivamente (1,9). La mortalidad general relacionada con la enfermedad oscila entre 0% y el 80%, en las lesiones del RP si el seguimiento clínico se prolonga por 10 ó 20 años, respectivamente (22). Es importante destacar que el TLA/LPS BD no metastatiza hasta que no presente desdiferenciación, la cual suele acontecer al cabo de 7-8 años del supuesto inicio del tumor (1,8).
2. Liposarcoma desdiferenciado (LPS DD)
Se define como una neoplasia adipocítica maligna que presenta transición de TLA/LPS BD a sarcoma no lipogénico de grado histológico variable, normalmente de varios milímetros de diámetro (3). La transición puede presentarse en el tumor primario (ab initio) (90%) o en la recidiva de un TLA (10%) (1,3,5,8,23,24).
El riesgo de desdiferenciación en los TLA/LPS BD se estima en el 10% y es mucho más acusado en las lesiones centrales, particularmente en RP, que en las superficiales; no obstante, es el tiempo de evolución, en vez de la localización, el factor más determinante.
Las técnicas de citogenética y biología molecular han venido a confirmar la estrecha asociación entre el TLA/LPS BD y el LPS DD ya que ambas lesiones presentan cromosomas en anillo y/o gigantes y dmin (3,8,17). Una peculiaridad de los LPS DD que los diferencia de los TLA/LPS BD es la existencia de múltiples clones anómalos, uno o varios con cromosomas gigantes o en anillo (25,26). Estudios mediante FISH e hibridación genómica comparada han demostrado amplificación del segmento 12q13-21 asociada a coamplificación de otras regiones (17,25).
El LPS DD se presenta en el mismo grupo etario que el TLA/LPS BD y las localizaciones preferentes son: RP, extremidades (EE), cordón espermático y mediastino. La proporción RP/EE es de 3:1. Es de destacar que aunque es posible la desdiferenciación de TLA/LPS BD superficiales esta, cuando se presenta, es excepcional (9,22).
Los LPS DD del RP se presentan como tumoraciones no dolorosas que causan distensión abdominal o compresión de vísceras adyacentes. En las extremidades se muestran como lesiones de crecimiento rápido reciente desarrolladas en el seno una masa previa estable (8).
Macroscópicamente son masas amarillas grandes, multinodulares, con áreas sólidas gris-marrón no lipogénicas (fig. 12). Con frecuencia se observan focos de necrosis y hemorragia. En ocasiones, las áreas desdiferenciadas ocupan la totalidad de la lesión. Esto se observa especialmente en aquellas resecciones parciales donde el cirujano interpreta como tumor sólo las áreas blancogrisáceas y no el tejido adiposo periférico (1,8) (fig. 13).
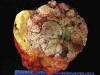
Fig. 12:
Liposarcoma desdiferenciado de retroperitoneo. Macroscopía. Lesión
característica donde existe transición brusca de áreas amarillentas (liposarcoma
bien diferenciado tipo lipoma) a áreas blanquecinas y necróticas, propias del
liposarcoma desdiferencado de alto grado.
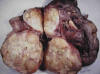
Fig. 13:
Liposarcoma desdiferenciado de retroperitoneo.
Macroscopía. Lesión multinodular de coloración blanquecina con pequeños focos de
hemorragia. No se reconocen áreas de tejido amarillento adiposo.
El cuadro histológico característico viene definido por la transición de un LPS BD a sarcoma no lipogénico que, en la mayoría de los casos, es de alto grado (fig. 14). Esta transición puede ser abrupta o progresiva; más raramente, se encuentran áreas interpuestas de sarcoma no lipogénico de bajo grado o bien la lesión se presenta como múltiples focos de desdiferenciación en el seno de un LPS BD (desdiferenciación en mosaico) (1,22,23). En muchas ocasiones el sarcoma no lipogénico se descubre en el lugar donde previamente había sido extirpado un TLA/LPS BD sin que exista transición entre una forma de tumor y otra (1,8) (fig. 13).

Fig. 14:
Liposarcoma desdiferencado de alto grado A.
Transición brusca entre liposarcoma bien diferencado tipo lipoma y liposarcoma
desdiferencado. HE 2,5x. B. Detalle del componente de liposarcoma bien
diferenciado con frecuentes células estromales atipicas. HE 40x. C. Detalle del
componente desdiferenciado de alto grado con marcada anaplasia celular. HE 20x.
En el curso de los últimos años han surgido varias cuestiones, aún no bien definidas, sobre los criterios diagnósticos del LPS DD.
¿Que cantidad de tejido desdiferenciado debe estar presente para cualificar a la lesión desdiferenciada?
¿Debe ser siempre el componente desdiferenciado de alto grado, o es posible encontrar áreas de bajo grado?
¿Deben incluirse en esta categoría a los tumores con diferenciación divergente?
Con respecto a la primera, debemos señalar que aunque la extensión del componente desdiferenciado puede ser muy variable, en la mayoría de los casos suele ser bien visible macroscópicamente; el componente mínimo necesario lo establecen algunos autores (8) en un campo microscópico de ¥10; no obstante, el significado pronóstico de estos focos microscópicos está aún por determinar.
En relación con el tipo de desdiferenciación (alto grado vs. bajo grado) existen ciertos desacuerdos. Inicialmente, Evans (22) aplicó el término de LPS DD a aquellos tumores que presentaban desdiferenciación de alto grado, de modo semejante a como Dahlin y Beaubout (27) lo habían aplicado en los condrosarcomas. En la actualidad la OMS (3), admite el concepto de desdiferenciación de bajo grado, que viene definida por la presencia de una proliferación fibroblástica con atipia leve, a menudo dispuesta en fascículos, y con una densidad celular intermedia entre LPS BD esclerosante y LPS DD de alto grado (1,23) (fig. 15). Las áreas de desdiferenciación de bajo grado no deben confundirse con el LPS BD tipo fusiforme, ya que este presenta en el seno de la celularidad fusiforme diferenciación adiposa (lipoblastos) (15). La desdiferenciación de bajo grado ha sido cuestionada por algunos autores (8) e interpretada como áreas celulares de LPS BD esclerosante; estos autores insisten en que para el diagnóstico de LPS DD es necesario, además del pleomorfismo nuclear y la ausencia de diferenciación lipogénica, un índice de mitosis Ž5 mitosis/10CGA.
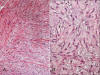
Fig. 15:
Lipoosarcoma desdiferenciado de bajo grado. A.
Área de aspecto tipo fibrosarcoma de bajo grado. HE 20x. B. Detalle de un área
con matriz mixoide y celularidad fusiforme tipo «cultivo celular». HE 40x.
Por último, sobre el patrón histológico de desdiferenciación insistir en la posibilidad de muy diversos cuadros histológicos que, a veces, pueden coincidir en una misma lesión. Las formas más habituales son las de tipo fibrohistiocitoma maligno pleomórfico (FHM) y mixofibrosarcoma de alto grado; menos frecuentemente se pueden encontrar áreas superponibles a otros sarcomas, tales como fibrosarcoma de alto grado, hemangiopericitoma, o bien cuadros morfológicos que con las técnicas de hematoxilona y eosina remedan otras neoplasias, como carcinomas poco diferenciados, melanomas, linfomas o lesiones de tipo meningotelial (1,3,5,8,22-24) (fig. 16). Estas últimas, con frecuencia se observan en asociación con metaplasia ósea madura. Diferenciación heteróloga propiamente dicha (osteosarcoma, condrosarcoma, leiomiosarcoma, rabdomiosarcoma y angiosarcoma) se ha observado en el 5-10% de los casos y carece de significación pronóstica (22).

Fig. 16:
Liposarcoma desdiferenciado de bajo grado con
áreas tipo meningotelial. A 10x. B 40x.
En general, no existe dificultad alguna para reconocer esta entidad cuando coexisten áreas de TLA/LPS BD y de sarcoma no lipogénico de alto grado. Los problemas se presentan cuando el componente de TLA/LPS BD es mínimo o no ha sido extirpado por el cirujano. El resultado es el de un diagnóstico de FHM o de otro tipo de sarcoma. En todos los tumores pleomórficos de alto grado situados en el RP debe buscarse de modo insistente la presencia de áreas de LPS BD, puesto que el pronóstico del LPS DD es mejor que el de otros sarcomas pleomórficos (23). Las técnicas de inmunohistoquímica pueden contribuir de modo importante en la resolución de esta cuestión, particularmente en los casos en que no se identifica componente de LPS BD, puesto que la co-expresión de MDM2 y CDK4 es muy común en los LPS DD y rara en sarcomas de otros tipos (24,28) (fig. 17). Mediante la técnica de FISH en el componente desdiferenciado de alto grado existe un aumento del número de copias centroméricas del cromosoma 12 (21), y de señales de amplificación de los genes MDM2 y CDK4, mucho más acusado que en los LPS BD/TLA (25) (fig. 11B). Aunque desconocemos los factores implicados en la evolución desfavorable de los sarcomas pleomórficos, algunos autores han sugerido la implicación del gen TP53, ya que su funcionalidad se encuentra más conservada en los LPS DD que en el resto de sarcomas pleomórficos (29).
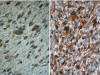
Fig. 17:
Liposarcoma desdiferenciado. Inmunoexpresión de
MDM2 (A) y de CDK4 (B). 40x.
La tasa de recidivas de los LPS DD se encuentra alrededor del 40% y es mucho mayor que la observada en los TLA/LPS BD (1,22,23). Es de destacar que prácticamente todos los LPS DD situados en el RP recidivan tras un seguimiento prolongado (19 o más años). Las metástasis a distancia se presentan en el 15-20% de los casos y suelen asentar en el pulmón, hígado y hueso, especialmente si el tumor es central (1,8). Histológicamente están constituidas exclusivamente por el componente desdiferenciado; esto no se sucede con las recidivas locales, donde a veces solo se observa componente de TLA/LPS BD, aunque lo habitual es que existan ambos componentes y en recidivas sucesivas vaya aumentado el componente no lipogénico (24).
La mortalidad media a los 5 años es del 18-30%, pero esta cifra alcanza hasta el 40% si el seguimiento se prolonga por 10-15 años (22).
El factor pronóstico más determinante es simplemente la localización, siendo peor en los de localización central –RP e ingle–; la extensión del componente desdiferenciado, el tipo de desdiferenciación o la presencia de metaplasias carecen de impacto pronóstico con significación estadística (1,22).
3. Liposarcoma mixoide (LPS MIX)
Tumor maligno compuesto por células mesenquimales uniformes, redondas u ovales, no lipogénicas, junto con un número variable de lipoblastos atípicos en el seno de un estroma mixoide con vascularización capilar plexiforme (3). Este término engloba las entidades LPS mixoide y LPS de células redondas de la anterior clasificación (4), puesto que en la actualidad estas se consideran extremos de un espectro lesional, con frecuentes formas transicionales y una única alteración cromosómica: la t(12;16)(q13;p11) o más raramente t(12;22)(q13;q12). Esta alteración da lugar a la formación de una proteína híbrida, resultante de la fusión del gen DDIT3 (CHOP) con el FUS (TLS) o EWS, según sea el tipo de traslocación (1,3,5,8,17). La fusión del gen DDTT3 (CHOP) es bastante característica del LPS MIX y no ha sido descrita en otros tipos de LPS o de tumores mixoides (3,30).
El LPS MIX representa el 30-35% de todos los LPS, muestra preferencia por adultos, con un pico de máxima incidencia entre la 4.ª-5.ª décadas de la vida, y asienta principalmente en las EEII, particularmente muslo, glúteo y hueco poplíteo. Rara vez se presenta en el tejido subcutáneo o en RP. Es de destacar que los casos descritos en la infancia son todos en niños mayores de 10 años (7,31).
Macroscopicamente son lesiones bien circunscritas, multinodulares e intramusculares, de color rojizo-tostado y consistencia gelatinosa (fig. 18). A veces, coexisten áreas amarillentas compuestas por adipocitos maduros o bien áreas con aspecto de carne de pescado, correspondientes a focos hipercelulares y de células redondas (1,3,5,8,31).
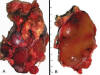
Fig. 18:
Liposarcoma mixoide. A. Lesión multinodular de
color rojo tostado con áreas adipocíticas más periféricas. B. Superficie de
corte que pone de manifiesto el aspecto gelatinoso.
El espectro histológico es bastante amplio (1,3,5,8,31). En un extremo tenemos el denominado LPS MIX clásico o paucicelular (8), compuesto por formaciones nodulares con escaso número de células, marcada trama vascular plexiforme (vasos capilares ramificados en ángulo recto) y abundante matriz mixoide. Las células proliferantes son pequeñas, fusiformes, y contienen escaso citoplasma y un núcleo oval con discreta hipercromasia (células indiferenciadas). Entremezcladas con estas células existen lipoblastos atípicos en diversos estadios madurativos (fig. 19). Los lipoblastos más maduros (lipocitos) tienen unas dimensiones menores que los adipocitos del tejido graso maduro y con frecuencia se disponen en la periferia de los nódulos tumorales y entorno a vasos (fig. 20). El número de lipoblastos atípicos y de células indiferenciadas varían notablemente de unos tumores a otros e, incluso en un mismo tumor, de unas áreas a otras (fig. 21). Las figuras de mitosis son muy ocasionales o incluso no llegan a identificarse (1,3,5,8,31). Con frecuencia, en el seno de la matriz mixoide se advierten formaciones microquísticas, a veces con formas bizarras o caprichosas, repletas de material mucoide; estos lagos mucoides son tan característicos que su ausencia obliga a revisar el diagnóstico de LPS MIX. (1,8) (fig. 22). Otros hallazgos menos constantes incluyen la presencia de hemorragia intersticial y focos de diferenciación heteróloga del tipo cartilaginosa, leiomiomatosa u ósea (32). Diferenciación rabdomiosarcomatosa, ha sido descrita en una ocasión (33) pero no es aceptada universalmente.
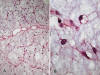
Fig. 19:
Liposarcoma mixoide. A. Lesión mixoide
paucicelular con marcada trama vascular plexiforme. HE 2,5x. B. Detalle donde se
advierten ocasionales lipoblastos atípicos multivacuolados y células
indiferenciadas. HE 60x.
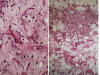
Fig. 20:
Liposarcoma mixoide. A. Detalle donde se
advierten todas las características propias de la lesión: matriz mixoide, patrón
vascular plexiforme, células indiferenciadas y lipoblastos atípicos. HE 40x. B.
Vista panorámica done se observa maduración adipocítica en la periferia de un
territorio con las características propias de liposarcoma mixoide. HE 2,5x.
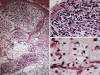
Fig. 21:
Liposarcoma mixoide. A. Imagen panorámica donde
se pone en evidencia la distribución irregular de la celularidad. HE 2,5x. B.
Detalle donde se advierten áreas hipercelulares con escasa diferenciación
lipoblástica. HE 40x. C. Área hipocelular dode se identifican lipoblastos
atípicos. HE 40x.
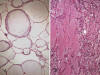
Fig. 22:
Liposarcoma mixoide. Lagos mucosos. A. Patrón
peculiar en el que la mayor parte de la matriz se dispone formando esferas con
distintas desnsidades. Las paredes están constituidas por las células tumorales
propiamente dichas. HE. 2,5x. B. En esta ocasión los lagos mucoides son más
irregulares, contactan entre sí y contiene un material denso y eosinófilo
remedando al edema de pulmón. HE. 2,5x.
En el otro extremo del espectro nos encontramos con lesiones muy celulares, LPS MIX celular (8), con escasa matriz mixoide intersticial, trama capilar poco manifiesta, frecuentes figuras de mitosis y células muy próximas entre sí y con tendencia a la superposición. Individualmente, las células presentan contorno circular y núcleos con cromatina grosera y nucléolos prominentes (fig. 23). Estas áreas, que son las que tradicionalmente se conocen como de células redondas (1), son muy poco frecuentes. Mucho más comunes son las denominadas áreas hipercelulares en las que la densidad celular es menor (ausencia de acumulaciones tridimensionales) y las células presentan una morfología semejante a la de las células indiferenciadas o bien son discretamente mayores (fig. 24). Tanto en el seno de las áreas de células redondas como hipercelulares con frecuencia se observan lipoblastos uni o multivaculados (1,35,8,31). Las áreas hipercelulares y de células redondas pueden observarse como fenómeno de desdiferenciación progresiva o transicional o bien como un nódulo aislado en el seno de una lesión paucicelular. Se denominan áreas transicionales a aquellas en las que hay aumento de la celularidad pero aún existe abundante matriz mixoide y conservación de la trama vascular plexiforme.
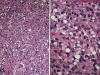
Fig. 23:
Liposarcoma de células redondas. A. Aspecto
panorámico donde se advierte una marcada densidad celular con muy escaso estroma
interpuesto y trama vascular poco relevante. HE. 10x. B. Detalle donde se
advierten numerosos lipoblastos univacuolados. Con frecuencia en los núcleos se
observan nucléolos prominentes. HE 40x.
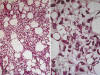
Fig. 24:
Liposarcoma mixoide con áreas hipercelulares. A.
En estos territorios la densidad celular es menor (ausencia de acumulaciones
tridimensionales) que la que se observa en los liposarcomas de células redondas.
HE, 10x. B. Las células presentan una morfología semejante a la de las células
indiferenciadas o bien son discretamente mayores. HE 40x.
Tomando en consideración que el pronóstico de los LPS MIX está íntimamente relacionado con la extensión de las áreas de células redondas, se ha prestado especial atención a su estratificación y se ha recomendado muestrear extensamente las lesiones (un bloque por cada cm. de diámetro máximo). Evans (34) ha distinguido tres grados con diferente evolución clínica: grado I (<5%); grado II (5-25%) y grado III (>25%) señalando notables diferencias pronósticas. Las lesiones grado I (que denomina LPS MIX) tienen buen pronóstico con una supervivencia a los 10 años que supera el 70%, a pesar de la elevada tasa de recidivas locales (50%). Las lesiones grado III (que denomina LPS de células redondas) se consideran sarcomas de alto grado y muestran una marcada capacidad metastatizante. En las lesiones grado II (LPS mixtos mixoides y de células redondas) la evolución es más difícil de predecir, pero algunos autores consideran que el 10% marca el límite entre las metastatizantes y no metastatizantes (5,8). En la serie publicada por Kilpatrick et al (31) además del porcentaje de células redondas (Ž5%) se seleccionó como factor predictivo la existencia de focos de necrosis que, por otro lado, son excepcionales en este tipo de lesiones.
Los principales problemas con que se encuentra este sistema de gradación son dos: por un lado, no tiene en cuenta el tamaño global de la lesión y, evidentemente, no contiene la misma masa tumoral un 5% de una lesión pequeña que de otra grande (1) y, por otro lado, en la práctica diaria resulta difícil diferenciar entre áreas hipercelulares y áreas de células redondas; para evitar este segundo problema, Fletcher (35) ha recomendado utilizar un punto de vista pragmático y las agrupa en un mismo parámetro.
Entre los marcadores inmunohistoquímicos y moleculares sólo la expresión de TP53 se correlaciona con la evolución de la enfermedad (36).
Los LPS MIX son lesiones prácticamente únicas en su capacidad para producir metástasis en los propios tejidos blandos (extremidad contralateral, axila, retroperitoneo) y hueso (predilección por vértebras) incluso antes de que aparezcan en el pulmón (37). Por otro lado, en un número importante de casos la enfermedad se presenta de modo sincrónico o metacrónico con varios focos tumorales. Este hecho, que inicialmente se interpretó como un fenómeno de multicentricidad tumoral, en la actualidad se considera siembra metastásica dado que en los distintos focos se ha encontrado un patrón clonal (8,30). De ahí, que los casos con presentación inicial en forma de múltiples focos evolucionen mal, con independencia del grado de celularidad.
Las recidivas locales son muy frecuentes (50%) (31), e independientes del grado tumoral (8), particularmente si el tratamiento quirúrgico no se acompaña de radioterapia. La tasa de metástasis oscila en torno al 35% con un intervalo medio largo, de 68 meses (36). La mortalidad está alrededor del 31% (31) pero los pacientes que fallecen por el tumor generalmente lo hacen muchos años después de descubrirse la enfermedad.
El diagnóstico diferencial incluye una amplia gama de lesiones, tales como neoplasias adipocíticas benignas (lipoblastoma y lipoma condroide), tumores mesenquimales mixoides benignos (mixoma intramuscular, angiomixoma agresivo) y malignos (mixofibrosarcoma, condrosarcoma mixoide, dermatofibrosarcoma protuberans mixoide), y tumores metastáticos (linfomas, carcinomas y melanomas) (1,5,8). En la mayoría de las ocasiones, el diagnóstico diferencial se plantea en casos extremos; es decir, en lesiones con muy escaso número de lipoblastos o bien con morfología de células redondas. En el primero de los casos, se debe considerar el diagnóstico de LPS MIX después de una búsqueda exhaustiva de lipoblastos y de descartar principalmente dos lesiones eminentemente mixoides: el mixoma intramuscular y el mixofibrosarcoma de bajo grado. En casos extremos, la técnica de FISH con sondas específicas para el cromosoma 12 puede ser determinante al identificar la ruptura del gen DDIT3 (CHOP) (fig. 25). En los LPS MIX con morfología de célula redonda el diagnóstico diferencial se plantea con carcinomas, linfomas y melanomas que contengan células en anillo de sello y se parezcan a lipoblastos univacuolados. En estos casos, las técnicas de inmunohistoquímica son de notable ayuda, ya que los lipoblastos son positivos a la proteína S-100 y en las otras variedades tumorales existe inmunoreactividad específica a la citoqueratina (carcinoma), antígeno H-MB45 (melanoma) y antígeno leucocitario común (linfoma) (1,8).

Fig. 25:
Liposarcoma mixoide. Sonda cromosómica que
permite identificar mediante FISH la traslocación DDTI3(CHOP)/EWS, propia del
liposarcoma mixoide. Las células que la presentan muestran una señal con
coloración roja y verde conjunta.
4. Liposarcoma pleomórfico (LPS PLEO)
Se define como un sarcoma pleomórfico de alto grado con variable proporción de lipoblastos pleomórficos (3). Representa el 10-15% de los LPS (1) y el 20% de los sarcomas pleomórficos (3). La mayoría de los casos se observan en adultos mayores (>50 años) sin predilección por el sexo (1,5,8). Las localizaciones más habituales son las extremidades, particularmente las inferiores, seguido del RP, área paratesticular, mediastino, cabeza, cavidad pélvica, y órbita (1,3,5,8). Aunque la mayoría tienen una ubicación profunda, a veces también se pueden observan en hipodermis o piel (3,5,8). La mayoría de los pacientes se presentan con una masa firme de crecimiento progresivo y reciente aparición.
Macroscópicamente son lesiones firmes, por lo general mayores de 10 cm, multinodulares y blanco-amarillentas, que contienen áreas mixoides o de necrosis (1,3,5,8).
Histológicamente pueden presentarse como lesiones bien circunscritas o con contorno infiltrante. Se reconocen dos formas histológicas bien definidas aunque relacionadas (1,3,5,8). En ambas se advierte un patrón de crecimiento desordenado con extremo pleomorfismo celular y frecuentes células gigantes bizarras. Las diferencias principales entre ambos patrones radican en el contenido citoplásmico de lípidos. El patrón histológico más común se asemeja al histiocitoma fibroso maligno, pero contiene lipoblastos gigantes con núcleos bizarros e hipercromáticos; en muchos de estos lipoblastos los citoplasmas son marcadamente acidófilos y contienen numerosos glóbulos hialinos, que representan lisosomas gigantes (fig. 26). El segundo patrón es menos común y recuerda a un carcinoma de suprarrenal; está constituido por un crecimiento en sábana de células gigantes pleomórficas (generalmente las más numerosas) y células más pequeñas mononucleares (fig. 27). En ambos tipos celulares con frecuencia se observan lipoblastos atípicos pleomórficos. Miettienen y Enzinger (38) han descrito una tercera variante denominada epiteliode, caracterizada por un crecimiento en sábana de células poligonales, de límites bien definidos, citoplasma eosinófilo y núcleo redondo u oval con nucléolo prominente. Estos nidos se encuentran delimitados por septos con capilares finos y en su interior es posible encontrar diferenciación lipogénica focal.
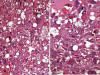
Fig. 26:
Liposarcoma pleomórfico. A. Sarcoma de alto grado
de malignidad con células gigantes pleomórficas y frecuentes lipoblastos
atípicos. Esta lesión tiene cierta similitud con Histiocitoma Fibroso Maligno el
HE 40x. B. Detalle donde se observan glóbulos hialinos extracelulares. HE 60x.
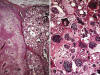
Fig. 27:
Liposarcoma pleomórfico. A. Imagen panorámica
donde se advierte una disposición en compartimentos. Alternan áreas de marcado
pleomorfismo nuclear con otras de aspecto más uniforme. HE 2,5x. B. Detalle
donde se advierte el pleomorfismo nuclear y el citoplasma con numerosas vacuolas
pequeñas que deforman el contorno nuclear. Esta lesión remeda a un carcinoma de
suprarrenal. HE 640x.
Las técnicas de inmunohistoquímica tienen escaso interés diagnóstico. En más del 50% de los casos existe inmunoreactividad específica a la proteína S-100 (3). Es de destacar que la variante epitelioide se pueden presentar marcadores epiteliales (38). Los estudios citogenéticos y moleculares realizados hasta la fecha son muy escasos y han revelado alteraciones complejas, no observadas en otras lesiones adipocíticas, que afectan principalmente a los cromosomas 8, 12 y 17, (17,39). Este hallazgo tiene notables interés patogenético puesto que apoya el concepto de independencia tumoral y va en contra de una progresión clonal anaplásica de otra forma bien definida de LPS.
Los LPS PLEO son neoplasias agresivas con una tasa de metástasis del 30-50% y de mortalidad del 40%. Las metástasis asientan preferentemente en el pulmón (1,3,8). En la serie publicada por Hornick y cols (40), que incluye a 57 pacientes, la supervivencia a los cinco años fue del 63%, con una tasa de recidivas locales y de metástasis del 42% y una supervivencia libre de enfermedad del 39%. En el estudio multivariable solo se seleccionaron como variables independientes predictivas de pronóstico adverso la edad (>60 años), la localización central, el tamaño tumoral y el índice de mitosis >10mitosis/ 10 campos de gran aumento.
5. Liposarcomas mixtos (LPS mixtos) e inclasificables
Son lesiones extremadamente raras que se observan en adultos de edad avanzada. Las combinaciones más comunes son de LPS MIX/ TLA/LPS DESD y LPS MIX/ LPS PLEO (3). Aunque la mayoría de los casos se han descrito en el RP o en la cavidad abdominal, también se han referido en otras localizaciones, tales como mediastino y extremidades (2,7). Macroscópicamente son lesiones grandes, multinodulares, con áreas sólidas gris-amarillentas, que suelen descubrirse de modo incidental (3).
Histológicamente, la mayoría corresponden a TLA/LPS BD con degeneración mixoide o desdiferenciación a mixofibrosarcoma. Otra segunda posibilidad es el desarrollo en un LPS MIX de áreas sarcomatosas no lipogénicas. Esta lesión también ha sido denominada LPS MIX desdiferenciado (41).
Los escasos estudios citogenéticos realizados corresponden a la primera categoría descrita y han revelado la presencia de cromosomas gigantes o en anillo del 12, hallazgos propios de los TLA/LPS BD/LPS DD. Esto obliga a considerar que las denominadas formas mixtas, al menos en parte, son formas inusuales de otras variedades de LPS (3).
BIBLIOGRAFÍA
Weiss SW, Goldblum JR. Enzinger and Weiss’s Soft Tissue Tumours. 4th ed. St Louis: Mosby; 2001. p. 571-694.
Dei Tos AP. Liposarcoma new entities and evolving concepts. Ann Diagn Pathol 2000; 4: 252-66.
Fletcher CDM, Unni KK, Mertens F (Eds): Adipocytic tumors. In World Health Organization Classification of tumors. Pathology and genetics of tumors of soft tissue and bone. Lyon: IARC Press; 2002. p. 19-46.
Weiss SW. Histological Typing of soft tissue tumours. En: Sobin LH (ed). World Health Organization Histological Classification of Tumours. Berlin: Springer Verlag; 1994.
Fletcher CDM. Soft tissue tumours. In: Fletcher CDM (ed). Diagnostic histopathology of tumours. Edinburgh: Chuchill Livingstone; 1995. p. 1474-85.
Mentzel T. Biological continuum of benign, atypical, and malignant mesenchymal neoplasms -does it exist? J Pathol 2000; 190: 523-5.
Shmookler BM, Enzinger FM. Liposarcoma occurring in children. An analysis of 17 cases and review of the literature. Cancer 1983; 52: 567-74.
Kempson R, Fletcher CDM, Evans H, Henrickson MR, Sibley RK. Lipomatous Tumors. In Tumors of the Soft Tissues. Atlas of Tumor Pathology. Third edition; n.º 30. Washington DC: AFIP; 2001. p. 187-237.
Weiss SW, Rao VK. Well-differentiated liposarcoma (atypical lipoma) of deep soft tissue of the extremities, retroperitoneum, and miscellaneous sites. A follow-up study of 92 cases with analysis of the incidence of «dedifferentiation». Am J Surg Pathol 1992; 16: 1051-8.
Evans HL, Soule EH, Winkelman RK. Atypical lipoma, atypical intramuscular lipoma, and well differentiated retroperitoneal liposarcoma: a reappraisal of 30 cases formely classified as well differentiated liposarcoma. Cancer 1979; 43: 1150.
Wenig BM, Heffner DK. Liposarcomas of the larynx and hypopharynx: a clinicopathologic study of eight new cases and a review of the literature. Laryngoscope 1995; 105: 747-56.
Austin RM, Dupree WB. Liposarcoma of the breast: a clinicopathologic study of 20 cases. Hum Pathol 1986; 17: 906-13.
Tallini G, Erlandson RA, Brennan MF, Woodruff JM. Divergent myosarcomatous differentiation in retroperitoneal liposarcoma. Am J Surg Pathol 1993; 17: 546-56.
Kraus MD, Guillou L, Fletcher CD. Well-differentiated inflammatory liposarcoma: an uncommon and easily overlooked variant of a common sarcoma. Am J Surg Pathol 1997; 21: 518-27.
Nascimento AF, Fletcher CDM. Spindle cell liposarcoma/Atypical lipomatous tumor. A clinicopathologic study of 120 cases. 94th Annual meeting. United States and Canadian Academy of Pathology. Modern Pathol 2005; 18 (suppl 1): 19A.
Pedeutour F, SuijkeRBuijk RF, Forus A, Van Gaal J, Van de Klundert W,Coindre JM, Nicolo G, Collin F, Van Haelst U, Huffermann K, y cols. Complex composition and co-amplification of SAS and MDM2 in ring and giant rod marker chromosomes in well-differentiated liposarcoma. Genes Chromosomes Cancer 1994; 10: 85-94.
Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: liposarcoma. Cancer Genet Cytogenet 2004; 155: 1-24.
Pilotti S, Della Torre G, Lavarino C, Sozzi G, Minoletti F, Vergani B, Azzarelli A, Rilke F, Pierotti MA. Molecular abnormalities in liposarcoma: role of MDM2 and CDK4-containing amplicons at 12q13-22. J Pathol 1998; 185: 188-90.
Dei Tos AP, Doglioni C, Piccinin S, Sciot R, Furlanetto A, Boiocchi M, Dal Cin P, Maestro R, Fletcher CD, Tallini G. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J Pathol 2000; 190: 531-6.
Pilotti S, Della Torre G, Mezzelani A, Tamborini E, Azzarelli A, Sozzi G,Pierotti MA. The expression of MDM2/CDK4 gene product in the differential diagnosis of well differentiated liposarcoma and large deep-seated lipoma. Br J Cancer 2000; 82: 1271-5.
Segura-Sánchez J, González-Cámpora R, Pareja-Megía MJ, García-Escudero A, Galera-Ruiz R, López-Beltrán A. Chromosome 12 copy number alterations and MDM2, CDK4 and TP53 expression in soft tissue liposarcoma. Anticancer Res (aceptado).
Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol 1979; 3: 507-23.
Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol 1997; 21: 271-81.
McCormick y cols D, Mentzel T, Beham A, Fletcher CD. Dedifferentiated liposarcoma. Clinicopathologic analysis of 32 cases suggesting a better prognostic subgroup among pleomorphic sarcomas. Am J Surg Pathol 1994; 18: 1213-23.
Meiss-Kindblom JM, Sjögren H, Kindblom LG, Peydro-Mellquist A, Roijer E, Aman P, Stenman G. Cytogenetic and molecular genetic analysis of liposarcomas and its soft tissue simulators: recognition of a new variants and differential diagnosis.Virchows Archiv 2001; 439: 141-51.
Mertens F, Fletcher CD,Dal Cin P, de Wever I, Ma ndahl N, Mitelman F, Rosai J, Rydholm A, Sciot R, Talini G, vam den Berghe H, Van R, Willén H. Cytogenetics análisis of 46 pleomorphic soff tissue sarcomas and correlation with morphologic and clinical features: a report of the CHAMP study group. Genes Chromosomes and Cancer 1998; 22: 16-25.
Dahlin DC, Beaubout JW. Deddiferentiation of low-grade chondrosarcoma. Cancer 1971; 28: 461-6.
Binh MB, Sastre-Garau X, Guillou L, de Pinieux G, Terrier P, Lagace R, Aurias A, Hostein I, Coindre JM. MDM2 and CDK4 immunostainings are useful adjuncts in diagnosing well-differentiated and dedifferentiated liposarcoma subtypes: a comparative analysis of 559 soft tissue neoplasms with genetic data. Am J Surg Pathol 2005; 29: 1340-7.
Dei Tos AP, Doglioni C, Piccinin S, Maestro R, Mentzel T, BaRBareschi M,Boiocchi M, Fletcher CD. Molecular abnormalities of the TP53 pathway in dedifferentiated liposarcoma. J Pathol 1997; 181: 8-13.
Antonescu CR, Elahi A, Healey JH, Brennan MF, Lui MY, Lewis J, Jhanwar SC, Woodruff JM, Ladanyi M. Monoclonality of multifocal myxoid liposarcoma: confirmation by analysis of TLS-CHOP or EWS-CHOP rearrangements. Clin Cancer Res 2000; 6: 2788-93.
Kilpatrick SE, Doyon J, Choong PF, Sim FH, Nascimento AG. The clinicopathologic spectrum of myxoid and round cell liposarcoma. A study of 95 cases. Cancer 1996; 77: 1450-8.
Siebert JD, Williams RP, Pulitzer DR. Myxoid liposarcoma with cartilaginous differentiation. Mod Pathol 1996; 9: 249-52.
Shanks JH, Banerjee SS, Eyden BP. Focal rhabdomyosarcomatous differentiation in primary liposarcoma. J Clin Pathol 1996; 49: 770-2.
Evans HL. Liposarcomas. A study of 55 cases with reassessment of its classification. Am J Surg Pathol 1979; 3: 507-23.
Fletcher CDM. Will we ever reliably predict prognosis in a patient with myxoid and round cell liposarcoma. Adv Anat Pathol 1997; 4: 108-13.
Schneider-Stock R, Ziegeler A, Haeckel C, Franke DS, Rys J, Roessner A. Prognostic relevance of TP53 alterations and Mib-1 proliferation index in subgroups of primary liposarcomas. Clin Cancer Res 1999; 5: 2830-5.
Spillane AJ, Fisher C, Thomas JM. Myxoid liposarcoma-the frequency and the natural history of nonpulmonary soft tissue metastases. Ann Surg Oncol 1999; 6: 389-394.
Miettinen M, Enzinger FM. Epithelioid variant of pleomorphic liposarcoma: a study of 12 cases of a distinctive variant of high-grade liposarcoma. Mod Pathol 1999; 12: 722-8.
Rubin BP, Fletcher CD. The cytogenetics of lipomatous tumours. Histopathology 1997; 30: 507-11.
Hornick JL, Bosenberg MW, Mentzel T, McMenamin ME, Oliveira AM, Fletcher CD. Pleomorphic liposarcoma: clinicopathologic analysis of 57 cases. Am J Surg Pathol 2004; 28: 1257-67.
Mentzel T, Fletcher CD. Dedifferentiated myxoid liposarcoma: a clinicopathological study suggesting a closer relationship between myxoid and well-differentiated liposarcoma. Histopathology 1997; 30: 457-63.
![]()