
Vol. 35, n.º 1, 2002
| REVISTA
ESPAÑOLA DE Vol. 35, n.º 1, 2002 |
Arantxa Guimerà, Xavier Gironès, Félix F. Cruz-Sánchez
Instituto de Ciencias Neurológicas y Gerontológicas, Universitat International de Catalunya
INTRODUCCIÓN
La enfermedad de Alzheimer (EA) esta considerada como la principal causa de demencia y ésta, es la cuarta causa de muerte en países desarrollados (1). Se define como un padecimiento neurodegenerativo del sistema nervioso central y se caracteriza por un deterioro progresivo de las funciones cerebrales superiores. Una consciencia notable del impacto social de la EA durante la última década ha llevado a grandes esfuerzos en la investigación con el fin de determinar la etiopatogenia, diagnóstico y tratamiento de esta enfermedad. Sin embargo, la causa de la EA no ha sido aún clarificada.
La demencia es un síndrome clínico, lo que implica que no existe una única explicación nosológica del mismo. La mayoría de los casos de EA son esporádicos, y un 5% tiene un patrón de herencia dominante (enfermedad de Alzheimer familiar: EAF).
Todos aquellos casos de edad avanzada (por encima de los 65 años) con demencia se engloban dentro de lo que se conoce como Demencia senil tipo Alzheimer (DSTA), ya que el sustrato morfológico puede ser diferente. De todas maneras, se acepta como definición dos grupos de EA según la edad de inicio del cuadro clínico:
Forma presenil o temprana (EA de inicio precoz): generalmente con agregación familiar; comienza antes de los 65 años de edad y constituye el 5 al 10% de todos los casos.
Forma senil o tardía (EA de inicio tardío): aparece después de los 65 años de edad; en su mayor parte esporádica, y representa entre el 90 y 95% de todos los casos.
Aún así, se discute si la EA de inicio precoz o la de inicio tardío se deben considerar como la misma enfermedad, aunque morfológicamente no presentan diferencias
En las formas familiares se han identificado diferentes genes cuyas mutaciones conducen a la acumulación del péptido
b-amiloide (Ab) involucrado en la fisiopatogenea de la enfermedad. Los genes descritos hasta el momento asociados como factor causal de la EAF son:Gen de la proteína precursora amiloide (PPA), localizado en el cromosoma 21.
Gen de la presenilina 1 (PS1), localizado en el cromosoma 14.
Gen de la presenilina 2 (PS2), localizado en el cromosoma 1.
Por otro lado, se ha descrito que el alelo (4 de la apolipoproteína E (ApoE), localizado en el cromosoma 19, es un potente factor de susceptibilidad para el desarrollo de la enfermedad de Alzheimer en la forma esporádica.
El incremento de la población de edad avanzada en los últimos años y el probable incremento en el futuro, ha hecho que aumente, y aumentará, la incidencia de demencia en este grupo poblacional. La ausencia de marcadores biológicos específicos para determinar los tipos de demencia hacen que el diagnóstico neuropatológico después de la muerte sea crucial para determinar el diagnóstico definitivo de un paciente que padece un síndrome neurologico de demencia.
NEUROPATOLOGÍA
PATOLOGÍA MACROSCÓPICA
Las alteraciones están tipificadas por atrofia generalmente simétrica y difusa de los giros cerebrales (figs.1 y 2), que se evidencia en la disminución del espesor de las circunvoluciones, aumento en la profundidad de los surcos, dilatación del sistema ventricular y disminución del peso y volumen cerebral (existe una correlación negativa entre el peso del encéfalo y el tiempo de evolución de la enfermedad). La atrofia levemente asimétrica es menos frecuente.
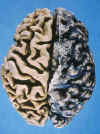
Fig. 1. Visión superior del
cerebro de un paciente afecto de EA. En el hemisferio izquierdo ha sido
eliminada la aracnoides y gran parte de los vasos que ocupan el espacio
subaractoideo. Nótese la marcada atrofia cerebral difusa que caracteriza a la
EA, el ensanchamiento de las fisuras y el adelgazamiento de las
circonvuliciones.
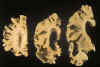
Fig. 2. Cortes coronales de un
hemisferio provinientes de un paciente afecto de Alzheimer. Nótese la marcada
atrofia cortical así como subcortical, puesta de manifiesto en la dilatación
de las cavidades ventriculares.
La atrofia afecta a los lóbulos temporales (más frecuentemente), frontales, parietales u occipitales. El patrón de atrofia más común es el difuso, seguido por una combinación de atrofia fronto-temporal, frontal o temporal aisladas, y en menor proporción puede haber un compromiso parieto-occipital.
Secciones a través de los hemisferios cerebrales revelan un adelgazamiento de la lámina cortical y dilatación simétrica del sistema ventricular (hidrocéfalo «ex-vacuo»). Los ganglios basales, diencéfalo, mesencéfalo y el tronco cerebral no muestran anormalidades notables. El cerebelo no muestra lesiones francas.
PATOLOGÍA MICROSCÓPICA
La patología microscópica incluye:
Placas seniles (PS), (difusas y clásicas).
Ovillos neurofibrilares (DNF).
Hilos del neurópilo (HN).
Pérdida neuronal y de sinapsis (degeneración neuronal).
Depósitos de amiloide en el cerebro y vasos sanguíneos cerebrales y meníngeos.
Degeneración gránulo-vacuolar en las células piramidales del hipocampo.
Presencia de cuerpos de Hirano.
Gliosis reactiva.
Aumento de las células de la microglía.
Alteraciones pseudo-espongiformes.
El hipocampo, el subiculum, la amígdala y las áreas de asociación neocorticales muestran las alteraciones más graves. El hipocampo y la corteza del lóbulo temporal están casi siempre afectados y muestran un patrón topográfico del progreso que fue utilizado para definir varias etapas histopatológicas tempranas y tardías de la EA (2). El núcleo basal de Meynert (área innominata) revela una predilección por la pérdida neuronal, formación de ovillos (degeneración neurofibrilar) y ausencia de placas seniles características.
Algunos estudios revelan que la progresión del deterioro cognitivo en la EA se deben principalmente a la pérdida de sinápsis, no encontrándose relación entre el número de placas seniles y el deterioro cognitivo.
Placas seniles (PSs)
Las placas son lesiones del neurópilo (entiéndase por neurópilo todo aquello que no es cuerpo celular ni vaso sanguíneo) de estructura esferoide que miden aproximadamente 20-100 µm de diámetro. Además de la proteína
b-amiloide, se han detectado muchas sustancias en PSs (fig. 3), incluyendo amiloide sérico P así como varias proteínas de fase aguda, factores de complemento, proteoglicanes, apolipoproteína e4, citoquinas y una proteína no caracterizada llamada NAC (componente no amiloide) que deriva de la sinucleina.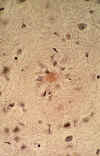
Fig. 3. Corte histológico teñido
con rojo congo que muestra en el centro de la imagen un depósito de substancia
amiloide rodeado de células de la microglía que constituye una placa senil
cortical (x400).
Se han descrito varios subtipos de placa en función del contenido relativo de amiloide, neuritas distróficas, células gliales o la presencia de capilar central (3,4,5,6,7):
Placas seniles difusas: Se la llama difusa por su apariencia poco demarcada. Están formadas por una delicada red de finas fibrillas de filamentos de amiloide, sin neuritas degeneradas ni zona central de amiloide condensado (fig. 4).
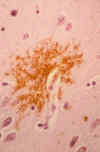
Fig. 4. Depósito de substancia
amiloide formada por una red de filamentos sin neuritas degeneradas ni zona
central de amiloide condensado.
Placas seniles primitivas: Están compuestas por depósitos extracelulares de Ab no fibrilar o escasamente fibrilar que la hace insoluble. Tienen una distribución más extensa de lo que se había apreciado inicialmente con tinciones convencionales de amiloide o de plata y son el subtipo más frecuente de placa. Este tipo de deposición temprana de Ab se da en ancianos sin clínica de EA y en pacientes con síndrome de Down. Las placas comienzan a aparecer en un neurópilo aparentemente normal y preceden el desarrollo de otros componentes de placa, tales como neuritas distróficas o células gliales reactivas. Por estos motivos, las placas primitivas representan los cambios morfológicos más tempranos en la secuencia patogénica de la EA (fig. 5).
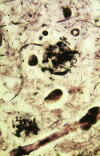
Fig. 5. Corte histológico teñido
con Bielschowsky que muestra placas seniles distróficas en el neurópilo
(x500).
Placas seniles clásicas (neuríticas): Están presentes en los estadios más avanzados de la enfermedad. Es un foco complejo de degeneración del neurópilo que contiene una región central de amiloide rodeada por astrocitos reactivos, microglía y neuritas distróficas que corresponden a dendritas y axones degenerados (fig. 6).
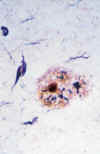
Fig. 6. Sección de tejido de un
caso de EA impregnada con un método de plata de Bielschowsky modificado. En el
centro se observa una placa neurítica. Esta es una lesión compleja que
contiene amiloide, procesos neuríticos distróficos y células gliales
reactivas. Obsérvese los procesos de tinción negros dentro de la placa. Estos
elementos son procesos cerebrales distróficos. Se requiere su identificación
para la clasificación de esta placa como neurítica. Esta placa en particular
exhibe una región central prominente de amiloide. Sin embargo, no se requiere
esta característica para clasificar esta placa como placa neurítica.
Obsérvese una DNF inmediatamente encima de la placa senil (ampliación x400).
Placas quemadas: Representan el estado terminal de la evolución de una placa particular en la que ha desaparecido el componente celular (no contienen neuritas anómalas asociadas). Sólo contienen una zona central de amiloide condensado.
Las PSs tanto clásicas como difusas y primitivas son más abundantes en la corteza, hipocampo y tálamo. En la corteza las placas se distribuyen más densamente en la base de los giros. En el cerebelo hay placas difusas y clásicas en todas las capas corticales.
Casi un tercio de los pacientes de edad avanzada (>74 años de edad) muestran formación extensa de placas con muy poca degeneración neurofibrilar (DNF). A estos casos se los ha denominado EA «placa predominante». Se ha postulado que esta variante representa un estadio en la enfermedad o alternativamente, que son ejemplos de una variante de la enfermedad que se conoce como enfermedad de cuerpos de Lewy difusos, una forma de demencia que puede iniciarse con trastornos motores como los de la enfermedad de Parkinson (EP), acompañados o seguidos de demencia o inversamente (8).
La disponibilidad de anticuerpos monoclonales que reconocen regiones específicas de la Ab demonstraron que los depósitos de amiloide en EA son idénticos a los que se producen en ancianos normales y pacientes con el síndrome de Down. Además estos depósitos no están limitados al cerebro humano y pueden producirse en otros mamíferos de edad avanzada y primates subhumanos (9).
Las PSs suelen ser más abundantes que los ovillos neurofibrilares (ONs), y más específicas de la EA, al contrario de las lesiones neurofibrilares, que parecen ser más el resultado de la muerte neuronal que una lesión primitiva de la célula (10). No obstante no se conoce bien la relación entre las PSs y la DNF. En el síndrome de Down primero aparecen depósitos difusos de Ab, a continuación las PSs y, por último, la DNF; mientras que en la EA el orden temporal acerca de cómo ocurre la degeneración permanece todavía sin dilucidar.
El examen con métodos combinados ha puesto de manifiesto que no existen neuritas distróficas aisladas; por el contrario, éstas siempre están asociadas a depósitos focales de amiloide (11). Estos datos sugieren un papel primario del amiloide en la génesis de las PSs (11). La formación y reclutamiento de neuritas distróficas alrededor de depósitos de amiloide es un fenómeno tardío.
Degeneración neurofibrilar (DNF)
La DNF está constituida por «filamentos helicoidales dobles» (FHD), los cuales están compuestos fundamentalmente por la proteína tau y neurofilamentos anormalmente fosforilados que corresponden a proteínas que forman parte del citoesqueleto neuronal normal (12) (figs. 7a y 7b). En el proceso de fosforilación anormal de la proteína tau y su consiguiente transformación en FHD intervienen dos enzimas (hiperactivación de una quinasa e hipoactivación de una fosforilasa). La proteína tau hiperfosforilada conduce a un ensamblaje y desensamblaje alterado de los microtúbulos, y también contribuye a una incorporación adicional de tau normal en filamentos anormales. La glicosilación no enzimática es otra vía que puede aumentar la fosforilación de tau anormal y la estabilización de filamentos ensamblados de forma anormal. Esta vía parece contribuir de forma importante a la insolubilidad de la DNF.
A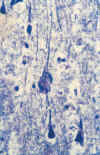
 B
B
Fig. 7. A) Neurona piramidal de la
corteza cerebral de un paciente con EA que muestra una banda filamentosa
marcadamente argirofílica en el citoplasma que rechaza al medio hacia la
periferia (x600). B) Degeneración neurofibrilar en neuronas de tamaño medio,
puesta de manifiesto con inmunotinción para la proteína tau (x250).
La formación de FHD genera como consecuencia una despolimerización de microtúbulos, lo cual da lugar a una alteración del transporte axonal, y consecuentemente a una disfunción y degeneración axonal. Los acúmulos de FHD bloquean el transporte de organelas y proteínas en el citoplasma neuronal, en los axones y dendritas, y la degeneración y muerte neuronal da lugar a una liberación de proteína tau a nivel extracelular y aparición en el líquido cefalorraquídeo.
Los ONs consisten pues en inclusiones filamentosas formadas dentro del citoplasma de las neuronas. Aunque las células piramidales son las que están principalmente implicadas, otros tipos de neuronas también pueden estar afectadas.
Las áreas predilectas de formación de DNF son el hipocampo, núcleo de Meynert, corteza cerebral y amígdala. Sin embargo, se puede encontrar DNF en otras regiones corticales y subcorticales, tales como la formación reticular, núcleos del rafe, mesencéfalo, locus coeruleus y núcleos pónticos. El ON puede asumir una forma de llama o globoide, y cuando se muere la neurona afectada, se transforma en una estructura eosinófila amorfa conocida como ovillo «fantasma». Ovillos intracelulares y extracelulares son fácilmente visualizados por varias tinciones histoquímicas para amiloide o por impregnación argéntica. La microscopía electrónica revela filamentos rectos y torcidos como los constituyentes más abundantes de la DNF. También un menor grado de estructuras membranosas y granulares contribuyen a su composición heterogénea. Los filamentos torcidos, también conocidos como filamentos helicoidales apareados, son el elemento estructural distintivo de la DNF. Las DNFs son relativamente insolubles. Muestran inmunoreactividad con anticuerpos policlonales y monoclonales dirigidos contra neurofilamentos y varios componentes de microtúbulos.
La DNF no es específica de la EA, sino que también se halla en el hipocampo de personas de edad avanzada y en numerosos trastornos neurodegenerativos como parkinsonismo postencefálico, encefalitis de cuerpos de inclusión, demencia pugilística, parálisis supranuclear progresiva (PSA), esclerosis lateral amiotrófica-Parkinson-demencia de Guam, Hallevorden-Spatz y en Nieman-Pick tipo C. Como estos trastornos no demuestran deposición amiloide, se ha propuesto que la DNF es una respuesta neuronal algo estereotípica al estrés neuronal.
La producción y acumulación de proteínas anormales como las observadas en la DNF en la EA induce la vía de degradación mediada por ubicuitina. La ubicuitina es una proteína de choque térmico de 8,6 KD, implicada en la degradación no lisosomal de proteínas anormales y otros mecanismos intracelulares proteolíticos. Cruz-Sánchez y cols. (12) demostraron que la ubicuitina se asocia de forma covalente con material neurofibrilar insoluble de la DNF en la EA. Además, existen diferencias topográficas en la distribución de la DNF en las diferentes poblaciones neuronales (incluidas las neuronas corticales) en la PSP y en la EA, y se ha demostrado (13) que se dan diferentes mecanismos físio- y etiopatogénicos en la producción de DNF en la EA y en la PSP. Estudios inmunohistológicos demostraron determinantes antigénicos diferentes en la DNF en la PSP y en la EA, basándose en la expresión de ubicuitina. La DNF cortical en la EA reflejó positividad con el antiserum anti-ubicuitina, mientras que la DNF en PSP fue negativa (12).
La ubicuitina tiene un papel en la deposición de amiloide en el cerebro, causando una deposición acelerada (14). La cosecreción de fragmentos peptídicos con ubicuitina debe ser considerado como un factor patogénico potencial en la formación de amiloide (15). Cruz-Sánchez y cols. (16) propusieron un posible papel para la ubicuitina en el proceso de deposición de amiloide en la angiopatía cerebral (AC). En la AC «pura» debe haber un proceso patológico primario que induce a la producción de ubicuitina, lo que debe conducir a la deposición de amiloide.
Demencia senil «DNF predominante»
Existe un subgrupo pequeño de pacientes afectados con demencia (aproximadamente un 5%), o sin ella, los cuales exhiben alteraciones neuropatológicas caracterizadas por un número importante de ONs en el hipocampo, pero sin PSs. Estos pacientes se clasifican como estadios límbicos avanzados en el sistema Braak & Braak, pero no cumplen con los requisitos para el diagnóstico de EA según el CERAD. La relación con la EA y la clasificación nosológica de esta variante no está resuelta aún.
La demencia «DNF predominante» afecta preferentemente a individuos de edad muy avanzada (>80 años) y tiene preferencia por las mujeres. Los casos típicos tienen afectación severa del hipocampo, amígdala, núcleo de Meynert y relativamene pocas alteraciones del neocortex.
La variante de la demencia con numerosos ONs debe distinguirse de otras condiciones con predominio de DNF. Estas incluyen la demencia fronto-temporal y las llamadas «tauopatías sistémicas múltiples» asociadas al cromosoma 17. Estas últimas se distinguen por una distribución de lesiones DNF e inclusiones gliales detectadas con inmunomarcación para tau con predominio fronto-temporal y subcortical.
Placas y DNF sin demencia
La «reserva» aparentemente grande del cerebro puede tolerar varios grados de formación de DNF o PSs con un compromiso funcional mínimo o indetectable. Ocasionalmente, individuos en la 8.ª y 9.ª década de vida sin demencia muestran densidades de DNF y placas similares a las diagnosticadas en la EA. Estas lesiones pueden haberse desarrollado a lo largo de periodos extensos, y los daños lentos habrían sido compensados por la «plasticidad» neuronal.
Otros cambios
Cambios en el Neurópilo
Los hilos del neurópilo (HNs) son estructuras filamentosas dispersas en el neurópilo, y ocurren independientemente de las PSs y DNF (17), aunque tienden a ser numerosas cuando hay presencia de ovillos.
Los HNs se desarrollan en las regiones alocorticales y isocorticales con una distribución y densidad variables en las diferentes áreas y capas corticales, siendo la lámina C cortical la más severamente afectada. Se ha sugerido que la acumulación de HNs en áreas corticales que contienen placas, depende de la cantidad de amiloide además de la DNF (18). Sin embargo, otros investigadores no han encontrado ninguna relación entre los HNs y las placas de amiloide (19). Los hilos neurofibrilares argirofílicos están prácticamente siempre presentes en la EA, y muestran propiedades inmunoquímicas y ultraestructurales idénticas a la DNF y procesos neuríticos distróficos. Estudios ultraestructurales de HNs han demostrado que éstos se componen de filamentos helicoidales apareados y filamentos rectos (20,21). Los HNs exhiben tinción positiva para anticuerpos anti-tau y anti-ubicuitina, así como para neurofilamentos fosforilados.
Angiopatía amiloide
En la mayoría de casos de EA, los depósitos de amiloide se encuentran también en los vasos sanguíneos (figs. 8a y 8b). La deposición de amiloide empieza en las capas externas musculares de arterias pequeñas y grandes. Fragmentos de PPA se depositan en la base de las membranas astrocíticas, miocíticas y pericíticas para formar fibrilas de amiloide típicas. Esta tendencia del amiloide a precipitar en la base de las membranas es una característica de todas las amiloidosis humanas.
A
 B
B
Fig. 8. Depósito de substancia de
amiloide en una arteriola intracerebral puesto de manifiesto por la
birrefringencia en la tinción de rojo congo. Nótese la coloración
amarillo-verdosa (manzana) del depósito amiloide (A) (x250) y arteriolas
subaracnoideas a través de inmunotinción para la proteína b-amiloide (A)
(x250).
El grado de angiopatía amiloide no se correlaciona ni con el grado de placas y ovillos, ni con el grado de deterioro cognitivo (22).
Degeneración granulo-vacuolar (DGV)
En la EA existe un hallazgo constante, pero no específico, caracterizado por la presencia de DGV en las células piramidales del hipocampo. Este cambio puede observarse fácilmente en preparaciones teñidas con hematoxilina-eosina y también con impregnación argéntica (figs. 9a y 9b). La mayoría de gránulos localizados centralmente son inmunoreactivos para ubicuitina (23). DGV se encuentra, fundamentalmente, en el sector C1 del hipocampo y en el subiculum, pero también puede hallarse en C2 y en la corteza entorrinal. Ocasionalmente, la degeneración neurofibrilar (DNF) y la degeneración granulovacuolar (DGV) ocurren en la misma célula (24).
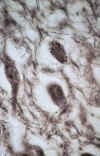
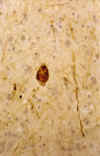
Fig. 9. Degeneración
gránulo-vacuolar en neuronas de tamaño medio vistas con impregnación
argéntica (A) e inmunotinción para neurofilamentos fosforilados (B) (x200).
Pérdida neuronal
La pérdida neuronal afecta particularmente a las capas superficiales de la corteza, y representa aproximadamente un 36% de la población neuronal. Las neuronas que sobreviven exhiben un nucleolo pequeño anormal y una reducción del RNA citoplasmático (25). Estudios comparativos de cromatina muestran un descenso en la cantidad de eucromatina con un incremento correspondiente de heterocromatina en neuronas y células gliales. Esto puede ser interpretado como una consecuencia de la reducción en la capacidad transcripcional (26).
Neurotransmisores
En el campo de los neurotransmisores, la característica más marcada es el descenso en la actividad colinérgica. La reducción en acetilcolintransferasa es el doble de la pérdida neuronal esperada en la corteza. Las terminales colinérgicas presinápticas están particularmente afectadas (27). También están afectadas las neuronas de proyección que producen transmisores monoamina, y neuronas corticales que producen glutamato, GABA, somatostatina, neuropéptido Y, factor liberador de corticotropina, substancia P, y otros neuromoduladores. Existe una reducción en la actividad de neurotransmisor en los sistemas serotoninérgico, noradrenérgico y dopaminérgico (28). No hay una correlación entre el grado de demencia y las anormalidades del sistema transmisor monoaminérgico. También se ha hallado reducciones en receptores muscarínicos en el hipocampo y receptores de GABA en el núcleo caudal (29).
Cuerpos de Hirano
Se encuentran fundamentalmente en el sector CA del hipocampo de un cerebro normal. En la EA son desplazados, frecuentemente, desde el estrato lacunar (su posición usual) hacia el estrato piramidal (fig. 10). Estos cuerpos contienen epítopos relacionados con microfilamentos, neurofilamentos y microtúbulos (30).
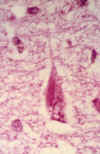
Fig. 10. Neurona piramidal que
muestra una banda filamentosa birrefringente en el citoplasma que corresponde a
un cuerpo de Hirano. Nótese además una degeneración gránulo-vacuolar en la
misma neurona (x600).
Procesos inflamatorios: activación microglial y macrofágica
Activación microglial
Uno de los procesos que contribuyen a exagerar la lesión neurodegenerativa es la reactividad microglial. Las células gliales regulan la actividad nerviosa ya que intervienen en fenómenos de plasticidad neuronal, supervivencia de las neuronas, nutrición neuronal, regulación del crecimiento, detoxificación y regulación homeostática (31).
Cuando el tejido nervioso sufre un daño, las células microgliales tienen la capacidad de actuar rápidamente y de manera heterogénea. Una vez activada, la microglía sufre cambios morfológicos, sobreexpresa diferentes receptores y es capaz de migrar, proliferar y transformarse en formas específicas de microglía reactiva. La reactividad de la microglía, mediante la fagocitosis y la secreción de elevados niveles de citocinas, proteasas y otros factores, puede cumplir una función destructiva o bien puede desempeñar una función beneficiosa (32,33).
La deposición de la proteína b-amiloide induce una respuesta inflamatoria local. También induce la activación de la microglía, mediante dos receptores microgliales: el receptor RAGE (Advanced glycation end products receptor) y el receptor SR (scavenger receptor). La microglía activada por los efectos del péptido b-amiloide experimenta una serie de cambios morfológicos asociados a un incremento de su metabolismo, expresión de novo de ciertas moléculas y sobreexpresión de otras ya presentes en la microglía inactiva. Esta actividad se ve asociada directamente al proceso lesivo neuronal por ser una gran fuente de radicales libres de oxígeno, contribuyendo a exagerar el estrés oxidativo y sus efectos neurodegenerativos.
La forma reactiva de la microglía es un estado muy complejo que incluye fagocitosis y también síntesis y expresión de diferentes moléculas involucradas en el control de la inflamación local y en la modulación de la respuesta inmunológica. La fagocitosis está protagonizada por macrófagos, que invaden el área lesionada para eliminar remanentes celulares. El origen de estos macrófagos puede derivar de la microglía residente o de la infiltración de monocitos exógenos sanguíneos. Estos últimos invaden el tejido nervioso atraídos por factores quimiotácticos, como la trombospondina y las b-quimiocinas, secretados por la microglía reactiva. La reacción inflamatoria protagonizada por la activación de la microglía viene asociada a una activación y proliferación de los macrófagos cerebrales.
Activación macrofágica
Dado que la EA está íntimamente asociada a la activación de macrófagos cerebrales residentes, se han evaluado los niveles de activación macrofágica en sangre periférica de enfermos de Alzheimer y el grado de apoptosis linfocítica. Se ha encontrado una correlación entre los fenómenos de activación macrofágica y apoptosis linfocitaria existente dependiendo del grado de la severidad clínica de la enfermedad. Se concluye entonces que el fenómeno de activación macrofágica periférica y apoptosis es un fenómeno paralelo al proceso cerebral inflamatorio en los enfermos de Alzheimer.
DIAGNÓSTICO
El diagnóstico de certeza de un síndrome demencial específico suele ser difícil de obtener durante la vida del paciente, por lo que es preciso efectuar un estudio anatomopatológico del tejido cerebral. Debido a la ausencia de marcadores específicos, el diagnóstico clínico del trastorno sigue siendo por lo tanto de exclusión. Los criterios expuestos en la cuarta edición del «Diagnostic and Statistical Manual of Mental Disorders of the American Psychiatric Association» (DSM-IV) (34) para el diagnóstico de la EA requiere el desarrollo de múltiples deficiencias cognitivas manifestadas tanto por: 1) deterioro de memoria, y 2) al menos una alteración cognitiva (afasia, apraxia, agnosia o alteración en funciones ejecutivas). También requiere los siguientes elementos:
A- Deterioro en desenvolvimiento social u
ocupacional y un declive de un nivel previo de la actividad habitual.
B- Inicio gradual y un declive cognitivo continuo.
C- Ausencia de otras condiciones que causan deficiencias progresivas de memoria
y cognitivas o que se conocen como una causa de demencia.
D- Las deficiencias no se desarrollan durante el curso del delirio.
E- El trastorno no está asociado a otra enfermedad neuropsiquiátrica.
No existen criterios de consenso universal para el diagnóstico de la EA. No obstante, muchos estudios y centros docentes han aceptado los criterios establecidos por «United States National Institute for Communicative Disorders and Stroke» y «The AlzheimerDisease and Related Disorders Association» (NINCDS-ADRDA) (35,36). Los criterios de NINCDS-ADRDA son similares y compatibles con los enumerados en la tercera edición del DSM (37), con la excepción de que no se requiere un deterioro en la actividad funcional diaria para el diagnóstico. El informe define demencia como «la disminución de memoria y otras funciones cognitivas comparadas al nivel previo de función del paciente, determinado por anormalidades observadas en examen clínico y pruebas neuropsicológicas.
LA NEUROIMAGEN EN EL DIAGNÓSTICO DE EA
La claridad diagnóstica del síndrome demencial se ha incrementado gracias a técnicas como la resonancia magnética (RM), porque provee más detalles anatómicos, mejor visualización de los cambios en la sustancia blanca subcortical y más opciones para planos en los cuales visualizar el cerebro con relación a la tomografía (TC) (38,39).
MÉTODOS DE DIAGNÓSTICO NEUROPATOLÓGICO
Métodos semicuantitativos:
CERAD (The Consortium to Establish a Registry for Alzehimer’s Disease) (40,41).
Khatchaturian (42).
Métodos topográficos como el de Braak & Braak (43), para determinar el estadio de severidad de la enfermedad.
Procedimientos ópticos estereológicos como el descrito por West (41,44).
Para realizar un diagnóstico definitivo de la EA es indispensable comprobar la presencia de PSs y ONs, dos hallazgos neuropatológicos clave, mediante un estudio anatomopatológico que demuestre estas lesiones en un paciente diagnosticado clínicamente de padecer Alzheimer. No obstante, es necesario la determinación de la existencia de PSs en una cantidad apreciable, puesto que, para algunos autores (45), en el envejecimiento normal existen PSs y ONs dispersos en la corteza, lo que para otros investigadores podrían ser casos «preclínicos» de EA (46).
Para identificar las lesiones neuropatológicas características de la EA se utilizan coloraciones histoquímicas basadas en plata tales como la de Bielschowsky, Holmes o Bodian, tioflavina S y Rojo Congo. La inmunohistoquímica demuestra varios componentes bioquímicos de las placas y ONs, mediante técnicas de inmunoperoxidasa con anticuerpos contra las proteínas b-amiloide, tau, neurofilamentos fosforilados y ubicuitina.
No hay un acuerdo universal en la comunidad neuropatológica sobre qué sistema debe usarse para el diagnóstico. La pérdida neuronal así como la reducción de la densidad sináptica no son cuantificados en un diagnóstico neuropatológico de rutina.
En los «Neuropathological Diagnostic Criteria for Brain Banking» (F.F.Cruz-Sánchez y cols., 1995) (47) se exponen los protocolos de diagnóstico actuales, así como los criterios de elección para el diagnóstico de EA.
PROTOCOLOS DE DIAGNÓSTICO
Los protocolos de diagnóstico para la EA tienen que considerar los siguientes aspectos:
1) Muestreo de tejido
Existen dificultades en la búsqueda de muestras cuando se recibe un cerebro que ya ha sido cortado en fragmentos irregulares y a menudo no muy identificables. Durante la búsqueda de muestra es importante obtener secciones transversales a través de planos rectos de la corteza. Si la evaluación microscópica (semi-cuantitativa o cuantitativa) se realiza con secciones oblícuas o tangentes, es probable que se cometan errores o estimaciones incorrectas de placas, ovillos y otras características de la patología de Alzheimer.
Hay una necesidad de obtener muestras de áreas múltiples del encéfalo. Estas muestras permiten realizar todas las observaciones CERAD, y hacer un diagnóstico de demencia que se podría confundir clínicamente con la EA.
Secciones adicionales del ganglio basal y del tálamo permiten la evaluación de la enfermedad cerebro-vascular y parálisis supranuclear progresiva.
2) Técnicas histológicas
Tinciones convencionales: Bielschowsky (figs. 5 y 6), Sevier-Munger, Gallyas, tioflavina S, rojo congo (figs. 3 y 8a), Golgi (48). Para evaluar la angiopatía amiloide se utiliza rojo congo y tioflavina S.
Inmunocitoquímica: anticuerpos contra tau, neurofilamentos, ubicuitina y b-amiloide (figs. 7b, 8b y 9b).
3) Criterios diagnósticos
Un buen método de diagnóstico tiene que ser:
a) Simple: Katchaturian y Tierney requieren contajes medios, mientras que CERAD realiza una estimación semi-cuantitativa de la implicación máxima. En la práctica, es más fácil la examinación del área de máxima implicación, mientras que para los valores medios han de ser examinados muchos más campos. Por tanto el CERAD ofrece una ventaja práctica en este sentido.
b) Transferible: El estándar tiene que poder usarse en diferentes centros y dar el mismo resultado.
c) Validado: El método propuesto debería tener una experiencia amplia internacional respecto a su uso. No sería apropiado proponer un protocolo completamente nuevo para el diagnóstico de la EA, pero sí sugerir la adopción de un diagnóstico estándar ya existente.
d) Versátil: El diagnóstico de la EA no se puede considerar aisladamente de otras enfermedades demenciales que se encuentran en un «banco de cerebros», lo cual se tiene que tener en cuenta en la proposición de un protocolo para el diagnóstico de la EA.
e) Comparable: Se tiene que realizar un test final para comparar los diferentes criterios de diagnóstico aplicados a un mismo grupo de pacientes. Katchaturian y CERAD proveen las mejores correlaciones, sobre todo CERAD.
CERAD
El CERAD ha realizado esfuerzos para estandarizar aún más las estrategias diagnósticas y de evaluación indicadas en el informe de la NINCDS-ADRDS.
El CERAD mide las manifestaciones cognitivas primarias de la EA, tales como el lenguaje, la memoria y la praxia a lo largo de un espectro de la gravedad del trastorno. Se diferencia bien entre los sujetos normales y los pacientes con demencia leve o moderada.
Los criterios establecidos por el CERAD y los de Khachaturian para el diagnóstico histopatológico se basan en un número específico de placas seniles relacionados con la edad y en un grado limitado de ONs en la neocorteza.
• Muestra de tejido:
1) Giro frontal medio (área 9).
2) Giro temporal superior y medio (áreas 21 y 22).
3) Lóbulo parietal inferior (áreas 39/40-partes 7 y 19).
4) Giro cingular anterior (área 24).
5) Hipocampo.
6) Amígdala y corteza entorrinal.
7) Parte media del cerebro, incluyendo la substancia negra.
Es posible la realización de secciones adicionales.
• Técnicas histológicas:
Se recomienda Bielschowsky, pero también se aceptan otras alternativas.
• Criterios de diagnóstico:
1) Evaluación de placas neuríticas.
2) Evaluación semi-cuantitativa de la implicación máxima.
3) Corteza frontal, temporal o parietal.
4) Recuento de placas relacionadas con la edad.
• Rangos: EA posible, probable o definida.
La actualización del método CERAD comporta una revisión mas detallada de la definición de EA posible y enfermedad de Parkinson, haciendo sugerencias específicas sobre las secciones tomadas para el diagnóstico de la demencia cerebrovascular.
Ningún protocolo de diagnóstico individualmente es completamente satisfactorio, pero CERAD tiene los méritos de ser:
— Relativamente simple.
— Aplicado exitosamente en un gran número de centros distintos.
— Reconocido ampliamente a nivel internacional y actualizado regularmente.
— Es un protocolo de diagnóstico apropiado para la mayoría de los casos de demencia.
KHATCHATURIAN
• Muestra de tejido:
1) 3 regiones de la neocorteza (frontal, temporal, parietal).
2) Hipocampo.
3) Amígdala.
4) Ganglio basal.
5) Substancia negra.
6) Corteza cerebral.
7) Médula espinal.
• Técnicas histológicas:
Coloración de plata de Bielschowsky, tioflavina S (con iluminación UV), rojo congo (con luz polarizada). No se recomienda tinción Bodian.
• Criterios de diagnóstico:
En las tres regiones de la neocorteza, en campo microscópico de 200 X:
1) Pacientes menores de 50 años: más de cinco PSs y ONs por campo de 200X.
2) Pacientes de edad entre 50 y 65 años: ocho o más PSs por campo de 200X, con o sin ONs.
3) Pacientes de edad entre 66 y 75 años: 10 o más PSs por campo de 200X, con o sin ONs.
4) Pacientes mayores de 76 años: más de 15 PSs por campo de 200X, con o sin ONs.
El recuento de las lesiones se realiza en las zonas donde se registra mayor densidad de ellas. El requerimiento del número de placas está reducido al 50% en pacientes con una historia de demencia. La presencia de otras enfermedades como la enfermedad de Parkinson y la enfermedad de Pick, también reduce el grado de cambios requeridos para el diagnóstico de EA.
Las diferencias entre CERAD y Khatchaturian son las siguientes:
1) Khatchaturian nos permite hacer un diagnóstico de la EA en ausencia de una historia de demencia y sin la presencia necesaria de ONs, particularmente en pacientes mayores de 75 años.
2) El CERAD presenta tres distinciones en el diagnóstico de la EA: posible, probable y definida, según el número de placas relacionadas con la edad examinadas de manera semi-cuantitativa, y la presencia o ausencia de una historia clínica de demencia. Usando los criterios CERAD en ausencia de una historia clínica no importa la severidad de la EA, sino que sólo se puede realizar un diagnóstico de la EA posible, lo cual concuerda con la proposición de que no hay criterios morfológicos absolutos para el diagnóstico de EA.
3) El CERAD se limita sólo a la cuantificación de las placas neuríticas, lo cual está de acuerdo con las estimaciones del número de placas realizadas en numerosas publicaciones que tratan de la gran importancia de la patología neurítica en los estudios clinico-patológicos relacionados con demencia (41).
4) Requerimiento por parte del CERAD del máximo grado de implicación en cada una de las áreas corticales (en lugar de una media de implicación). En la práctica ello conlleva una más fácil interpretación.
TIERNEY
• Muestra de tejido:
1) Frontal (5X), temporal (5X), parietal (3X), occipital (1X).
2) Alocorteza ( incluido el hipocampo) (3X).
3) Amígdala.
4) Ganglio basal, tálamo y núcleo basal.
5) Mesencéfalo, médula pons (3X).
6) Cerebelo.
• Técnica histológica:
Bielschowsky.
• Criterios diagnósticos:
A1) Uno o más ONs y una o más placas neuríticas en un campo de 25X en el hipocampo (ignorando los hallazgos neocorticales).
A2) Uno o más ONs y una o más placas neuríticas en un campo de 25X en la neocorteza y en el hipocampo.
A3) Uno o más ONs y una o más placas neuríticas en un campo de 25X en la neocorteza, ignorando hallazgos en el hipocampo. Además, los criterios de exclusión vascular: V1, V2, V3.
BRAAK y BRAAK
• Muestra de tejido:
— Hipocampo anterior.
— Hipocampo posterior.
— Occipital.
— Técnicas histológicas:
Secciones de 100 µm con Gallyas.
• Criterios de separación en etapas:
Semi-cuantitativo (+/+++) de ONs y HNs.
Los estadios definidos por este sistema abarca las fases preclínicas y sintomáticas, la acumulación progresiva de placas y ONs a lo largo del tiempo, y su diseminación extensa y progresiva a través del cerebro.
Braak y Braak describieron 6 estadios en la neuropatología de la EA (43) (fig. 11). Las células de proyección específica de la región transentorrinall perialocortical, que se localizan en las profundidades del surco rinal, son las primeras neuronas corticales que manifiestan los cambios (estadio I clínicamente silencioso).

Fig. 11. Desarrollo de los cambios
neurofibrilares desde el estadio I hasta el estadio VI en la formación
hipocampal, regiones entorrinal y transentorrinal y la neocorteza temporal. U:
Uncus; Pa: Parasubiculum; Pr: Presubiculum; S: Subiculum; RE: Región
Entorrinall; RT: Región Transentorrinalll; IT: Isocorteza Temporal; CA1: Primer
Sector de la Callosidad del Cuerpo Ammon. (Modificado de Braak y cols. 1995).
Los casos de estadio II muestran numerosos ovillos neurofibrilares y hilos del neurópilo en la región transentorrinall, y algunos adicionales en la región entorrinal. La destrucción cortical en el estadio II apenas impide la transmisión de la información neocortical (a través de la región entorrinal) a la formación hipocampal, pero sin exceder del umbral sobre el cual aparecen los síntomas clínicos iniciales. Algunos individuos desarrollan ONs/HNs iniciales a una edad sorprendentemente joven. Obviamente, la edad avanzada no es un prerrequisito para el desarrollo de la patología intraneuronal. Esta obsevación pone en duda teorías que intentan explicar los cambios como consecuencia de influencias nocivas que se espera que tengan efecto generalmente en la edad avanzada (estrés peroxidativo, disfunción mitocondrial, desequilibrio del metabolismo de la glucosa). Esto no excluye la posibilidad de que el estrés peroxidativo pueda contribuir a los cambios en los estadios avanzados de la enfermedad o influencie en el proceso patológico. Posiblemente no parece ser un factor primario en la patogénesis de las lesiones iniciales en la EA.
En los casos de estadios límbicos III o IV, la destrucción cortical es ya severa, pero limitada a unas pocas regiones alocorticales y áreas adyacentes. La característica clave de estos estadios es la notable destrucción de las capas entorrinals responsables de la transmisión de datos desde la neocorteza hasta el hipocampo y viceversa. Inicialmente, la formación hipocampal se ve afectada sólo ligeramente. En el estadio IV el proceso de destrucción se difunde desde la región entorrinal hacia la amígdala, el hipocampo y especialmente hacia las áreas de asociación de la neocorteza temporal basal. Los protocolos clínicos de muchos individuos en el estadio III o IV registran un deterioro de las funciones cognitivas y presencia de cambios sutiles de la personalidad. En otros individuos, la aparición de síntomas está todavía camuflada por capacidades de reserva del individuo, las cuales compensan la destrucción local. A causa de la expresión ocasional de los síntomas iniciales y las lesiones cerebrales características, los casos de estadios III y IV, se considera que representan la EA incipiente.
Los estadios neocorticales finales muestran una gran cantidad de ONs y HNs en cada subdivisión de la corteza cerebral. Una característica propia del estadio V es la destrucción extremamente severa de las áreas asociativas neocorticales, dejando solamente poco o nada afectados a los campos motores primarios, las áreas sensoriales primarias y sus regiones circundantes.
En el estadio VI, el proceso patológico se extiende a las áreas primarias. Los individuos caracterizados en los estadios V y VI de destrucción cerebral son todos dementes. Estos estadios corresponden a la EA completamente desarrollada.
La secuencia y el patrón de destrucción son muy similares al progreso de mielinización cortical en el desarrollo inicial, pero en orden contrario.
Diagnóstico Diferencial
Aspectos a tratar dentro del diagnóstico diferencial (DD):
1) DD de pacientes con demencia que están
sometidos a exámenes neuropatológicos con un diagnóstico clínico de EA.
2) Enfermedades que podrían confundirse histopatológicamente con EA, como por
ejemplo:
* El caso de la formación de inclusiones de tau fosforilada en neuronas vulnerables en la EA, donde la DNF es característica pero no específica, dado que también sucede en otros desórdenes neurodegenerativos llamados colectivamente «tauopatías» (49). En las tauopatías, la deposición de tau difiere en el patrón de distribución y en el tipo de células implicadas, aunque existen patrones de distribución de tau en algunas tauopatías similares al patrón que se da en la EA (particularmente en las partes superiores y dorsales). Este es el caso de la Parálisis supranuclear progresiva (PSP), que afecta particularmente al subtálamo y al núcleo dentado, encontrándose la DNF, mayoritariamente, en el tronco cerebral. Cuando afecta a la corteza cerebral lo hace en neuronas grandes de capas profundas (V y VI), en cambio en la EA la DNF afecta a las neuronas medianas en capas más superficiales (III y V). En la PSP existe también un acúmulo de neurofilamentos y tau1, pero no de tau2, ALZ50 y ubicuitina (encontrados en EA).
* La Enfermedad de Pick (EP): descrita primeramente por Pick (50) como una forma especial de atrofia cerebral. La EA y la EP tienen mucho en común, ocurriendo ambas, en algunos casos, en el mismo individuo (51,52). Además, comparten antígenos específicos de las neuronas degeneradas (53). Esto debe reflejar un defecto metabólico común en la formación de estos dos tipos de inclusiones intraneuronales (53).
* La Angiopatía congofílica o angiopatía amiloide cerebral (AAC), proceso de etiología desconocida caracterizado por una deposición de amiloide en la pared de pequeños vasos sanguíneos cerebrales y meníngeos. En ambas entidades hay PSs, DNF y acúmulo de sustancia amiloide en la pared de los vasos. Sin embargo, se ha comprobado (14) que la ubicuitina no se une a los depósitos de amiloide de vasos intercerebrales o leptomeníngeos en la EA. La AAC es característica de la EA, y también se encuentra en algunos ancianos. Se ha demostrado un incremento en la frecuencia del alelo T del gen de la alfa-1-antiquimotripsina en la AAC. Por otro lado, no se ha encontrado ninguna asociación entre alfa-1-antiquimotripsina y ApoE en EA y en Demencia vascular.
* El Síndrome de Gerstmann-Straussler (SGS), donde los depósitos de amiloide neocortical se acompañan de la formación de ovillos, aunque la diferente morfología de los depósitos de amiloide en SGS, con grandes y complejos depósitos, serían suficiente para sugerir el diagnóstico de SGS en vez de EA. Además, la profusión de las placas de amiloide en la capa molecular del cerebelo, característica de SGS, es bastante diferente de los hallazgos encontrados en EA. En casos de duda, la inmunocitoquímica demostraría la proteína priónica en los depósitos de amiloide en SGS.
* Terry y cols. (54) describieron EA sin DNF neocortical. Las PSs eran abundantes, y la DNF estaba presente en la formación hipocampal. Este patrón se encontró en un 30% de 60 casos de SDTA sobre una edad media de 74 años. En un estudio posterior (18) se mostró que en absencia de DNF neocortical, las neuritas de las placas no contienen filamentos helicoidales dobles ni proteína tau.
* Hansen y cols. (55) encontraron 5 casos de EA concominante con la Enfermedad de Cuerpos de Lewy difusos, con coexistencia de cambio espongiforme en el neurópilo. Muchos autores han descrito una combinación de patología tipo Alzheimer y Cuerpos de Lewy en individuos dementes. Muchos de estos casos cumplen los criterios histopatológicos de la EA, pero en otros, el número de PSs es insuficiente para certificar el diagnóstico de EA, y ocasionalmente hay demencia en absencia de cambios tipo Alzheimer, habiendo numerosos cuerpos de Lewy corticales. Los casos de EA coincidentes con numerosos Cuerpos de Lewy corticales se han clasificado como Variante de EA con Cuerpos de Lewy, una forma combinada de Alzheimer y Parkinson, donde los pacientes presentan, a menudo, características clínicas de parkinsonismo, tales como hipomimia, temblor en reposo, bradiquinesia, rigidez de cuello, y enlentecimiento de movimientos alternos rápidos. Por lo general, no mejoran con terapia levodopa.
* Braak y Braak (56) describieron en muchos casos de demencia, granos argirofílicos dispersos por el neurópilo, y cuerpos enrroscados de filamentos teñidos con plata, principalmente localizados en la sustancia blanca, cerca de la sustancia gris. Los cambios parecen estar asociados con la enfermedad de Parkinson y la EA. Posiblemente, algunos casos mostraron exclusivamente granos argirofílicos y cuerpos enrrollados, y fueron considerados el sustrato morfológico de una forma de demencia de inicio tardío diferente a la EA. Sin embargo, algunos de los casos presentados por Braak y Braak consistían en EA que además mostraban cuerpos fibrilares argirofílicos.
* Mizutani y cols. (57) describieron 7 casos de SDTA con hallazgos clínicopatológicos inusuales. La alteración mental empezó después de los 65 años en todos los pacientes. La característica clínica principal fue un cambio marcado de personalidad, además de una alteración de la función cognitiva. Estos pacientes mostraron una pérdida neuronal en un patrón laminar, con gliosis confinada en la región CA1 del hipocampo, el área del giro hipocampal (corteza entorrinal), y la corteza occipitotemporal media. La substancia blanca subcortical mostró más gliosis fibrilar que pérdida de mielina. DNF y PSs en la EA eran menores que en SDTA.
DIAGNÓSTICO BIOLÓGICO DE LA EA
Actualmente, no se dispone todavía de ningún instrumento diagnóstico escasamente invasivo y con adecuada sensibilidad, especificidad y valor predictivo, si exceptuamos la determinación de mutaciones genéticas en el reducido número de casos de EA de aparición precoz y herencia autosómica dominante, aunque gran parte de los esfuerzos actuales en el campo de la investigación de la EA plantean la necesidad de identificar precoz e inequívocamente la EA de otros procesos demenciales y, si fuera posible, discernir diferentes subgrupos dentro de la enfermedad (58).
Los marcadores biológicos de mayor relevancia y disponibles en la práctica hospitalaria son:
Marcadores determinantes de la enfermedad:
Mutaciones del gen de la PPA.
Mutaciones del gen de la PS 1.
Mutaciones del gen de la PS 2.
Marcadores de susceptibilidad:
ApoE.
Gen de la alfa-1-antiquimiotripsina.
Cromosoma 12.
Gen DLST en cromosoma 14.
DNA mitocondrial.
Marcadores fisiopatológicos:
Proteína tau.
b-amiloide.
Aspartatoaminotransferasa.
Determinación combinada tau/b-amiloide
Determinación combinada tau/aspartato.
Marcadores genéticos
La determinación de mutaciones genéticas está justificada en las formas familiares de EA de presentación precoz y herencia autosómica dominante cuando existen síntomas de la enfermedad. En familiares asintomáticos es opcional y dependerá de los deseos del individuo.
El valor diagnóstico del genotipo ApoE pone de manifiesto que:
1) Los criterios clínicos solos presentan una alta sensibilidad, pero un número elevado de falsos positivos (baja especificidad).
2) El uso secuencial de ApoE con los criterios clínicos proporciona un aumento considerable de la especificidad a costa de un descenso de la sensibilidad.
3) La determinación del genotipo ApoE estaría indicada sólo en aquellos casos en los que se cumplen los criterios clínicos de EA probable.
Marcadores bioquímicos
Existen anticuerpos neuronales capaces de detectar tau en líquido cefalorraquideo (LCR). Los pacientes con EA muestran una elevación de los niveles de tau en LCR con respecto a los controles. Los niveles de tau en EA no guardan relación con la edad, sexo, edad de comienzo o tiempo de evolución del proceso, ni con el grado de demencia, pero sí que existe relación entre niveles de tau y grado de deterioro.
La gran dispersión de valores de tau podría estar en relación con la heterogeneidad de la enfermedad, y también con el hecho de que la tau cuantificada corresponde a tau total, fosforilada y no fosforilada, sin distinción de las diferentes isoformas de esta proteína. Es importante destacar que pueden detectarse niveles de tau elevados en otras patologías como meningitis, infiltraciones meningeas, AVC, demencia con cuerpos de Lewy difusos, demencias frontales y Creutzfeldt-Jacob.
En condiciones normales puede detectarse en sangre una pequeña cantidad de b-amiloide-42. En los casos de mutaciones en PS 1, PS 2 y PPA los niveles sanguíneos están elevados con respecto a los pacientes con formas esporádicas de EA (que presentan niveles sanguíneos similares a los de los sujetos normales). Aunque potencialmente útil en casos familiares de EA relacionados con las mutaciones en PS 1, PS 2 y PPA, la determinación de b-amiloide-42 en plasma carece de utilidad diagnóstica en las formas esporádicas de la enfermedad.
Estudios para detectar depósito de b-amiloide en piel, vasos e intestino de pacientes con EA, no tiene aplicación clínica por existir también depósitos similares en algunos casos de personas de edad sin demencia.
En LCR es posible detectar b-amiloide total (b-amiloide-40 y b-amiloide-42), el cual es similar en EA y controles. En general no se evidencian diferencias significativas entre controles y EA con relación a la b-amiloide-40, pero sí se encuentra un descenso de b-amiloide-42 en la EA con respecto a los controles. El descenso de los niveles de b-42 en LCR en los pacientes con EA resulta paradójico si se tiene en cuenta que el depósito anormal de esta amiloide en las placas sería el causante de la enfermedad. Se especula con el hecho de que sería este depósito el que originaría una menor cantidad de b-amiloide-42 libre en LCR por el secuestro de la misma en las placas. En los estadios iniciales de la enfermedad los niveles de b-amiloide-42 están elevados en LCR y la severidad de la demencia se correlaciona con el descenso de la b-amiloide-42.
Tau y b-amiloide combinadas
La determinación combinada de tau y b-amiloide-42 en LCR proporciona cifras de sensibilidad y especificidad superiores al 80%, lo que convierte a esta prueba en un instrumento de potencial utilidad para discriminar la EA de otros procesos neurológicos y del envejecimiento normal.
Aspartatoaminotransferasa (AAT)
La alteración en el metabolismo de la glucosa a nivel cerebral en los pacientes con EA plantea la posible utilización de aminoácidos glucogénicos como fuente alternativa de energía, que daría lugar a un incremento de actividad de la encima AAT. La AAT está significativamente elevada en pacientes con EA respecto a los controles. Combinando la determinación de tau y ATT se encuentra una fuerte correlación entre la concentración de tau y la actividad de AAT en LCR.
MECANISMOS DE PRODUCCIÓN DE LA EA
Actualmente se desconoce la etiología exacta de la EA, e intentos de obtener información en esta dirección a partir de estudios epidemiológicos y clínicos, están enfocados en los análisis de factores de riesgo, entre los cuales la edad y los antecedentes familiares de demencia son los más fuertemente asociados a la EA (1). La causa de la EA es compleja y existe una visión prevalente que apoya mecanismos más complejos que engloban factores genéticos y ambientales (59). Esto resulta verdadero en los casos de EA de inicio tardío, pero no existe un acuerdo sobre el espectro del riesgo que conlleva tener un familiar de primer grado afectado por la EA de inicio tardío (59,60).
Se han identificado diversos loci que confieren susceptibilidad hereditaria, cuyas mutaciones conducen a la acumulación de b-amiloide anormal, PPA, PS1, PS2, y un factor de susceptibilidad, ApoE.
Factores adicionales que pueden tener un papel en la patogenia de la EA incluyen agentes infecciosos no identificados, edad de los padres, pacientes que recibieron circulación extracorpórea, trauma craneal, disfunción tiroidea, adicción al tabaco, educación y exposición al aluminio. Estudios epidemiológicos y clínicos de estos factores demostraron datos muy conflictivos, y su papel continúa siendo un tema de debate. Sin lugar a duda, un cuadro más completo y exacto surgirá con la identificación de más genes y factores de riesgo de la EA (1).
GENES ASOCIADOS A LA EA
Proteína precursora amiloide (PPA) y péptido b-amiloide (Ab)
El gen de la PPA se localiza en el brazo largo del cromosoma 21. Mutaciones en el gen de la PPA solamente representan un pequeño subgrupo de casos de EAF. Se han descrito unas seis mutaciones aproximadamente. La edad de inicio se encuentra entre los 39 y 67 años. Su herencia es autosómica dominante, con penetrancia completa, lo que implica que aquellos miembros portadores desarrollaran la enfermedad siempre y cuando alcancen la edad de riesgo.
A partir de la PPA se origina el péptido b-amiloide (de 40-43 aminoácidos), el cual se deposita en la EA en forma de fibrillas amiloideas en las PSs y en los vasos meníngeos y cerebrales (61). Por lo tanto, el Ab es un marcador patológico esencial del trastorno. También se producen pequeñas cantidades de Ab durante el metabolismo celular normal, y la identificación de tales fracciones ha abierto nuevas explicaciones alternativas sobre la acumulación del amiloide.
La cuestión de si el depósito de Ab desencadena, o sigue, alteraciones que culminarían con la muerte de neuronas es un punto fundamental, aunque controvertido.
La asociación entre la distribución topográfica de amiloide y la DNF es sólo parcial, porque se puede encontrar DNF en áreas libres de amiloide. Se ha discutido si el depósito de sustancia amiloide causa la EA o es sólo un marcador de este trastorno (62). No obstante, varias líneas de investigación apoyan el papel causativo del amiloide en la EA. La evidencia más importante en esta dirección resulta de estudios genéticos que demuestran una relación entre varias familias de EAF y varias mutaciones en el gen de PPA (63). Sin embargo, la manera en la cual estas mutaciones causan la acumulación de Ab no ha sido elucidada.
La mayoría de las mutaciones EAF están en la porción transmembrana de la PPA. De manera análoga, una mutación en el dominio hidrofóbico de la proteína codificada por el gen mec-4 causa degeneración neuronal lenta en C. elegans (64). Esto indica que mutaciones en ciertas regiones de proteínas pueden causar degeneración neuronal.
Otra evidencia importante apoyando el papel del amiloide en EA surge de la observación de que el depósito de amiloide preceda al desarrollo de alteraciones neurofibrilares (65). Está demostrado que la Ab es tóxica para las neuronas (66). Las propiedades citotóxicas de Ab están mediadas por radicales libres de oxígeno y también dependen de varios factores adicionales (estructura secundaria del péptido, estado de agregación, tiempo de exposición, osmolaridad, ph y concentración). No se conoce por completo los mecanismos de toxicidad, pero parece que están implicados en la oxidación directa de varios componentes celulares (p. ej.: oxidación de bombas de Ca2+ en la membrana, DNA, etc), un aumento de la sensibilidad a la excitotoxicidad o disturbios en la homeostasis del calcio. La evidencia in vitro de citotoxicidad está ahora apoyada por los nuevos modelos transgénicos desarrollados de EA en los cuales la sobreexpresión de un transgen PPA mutado causa alteraciones similares a placas seniles seguidas por degeneración neurítica. Parece así que la Ab contribuye de manera directa o indirecta a la degeneración y muerte neuronal observada en cerebros con EA.
Presenilinas
Las proteínas PS 1 y PS 2 codifican proteínas de alta homología y sus funciones aún se desconocen; pueden incluir la regulación de la apoptosis, destino celular y transporte citoplasmático.
PS 1
Está localizada en el cromosoma 14 y se han identificado unas 50 mutaciones. En la mutación E280A hay una sustitución de ácido glutámico por alanina en el codón 280 del gen de PS1 (38). Esta sustitución determina un aumento de los depósitos de b-amiloide en la corteza cerebral, con predominio de b-amiloide de 42 aminoácidos. Su herencia es autosómica dominante. Éste se ha constituido en el grupo familiar más numeroso del mundo afectado por EA de inicio temprano con una causa etiopatogénica homogénea. El riesgo de padecer la enfermedad es de un 100% puesto que la existencia de la mutación se correlaciona en un 100% con el desarrollo de la enfermedad.
PS 2
Localizada en el cromosoma 1 y presente en formas familiares de inicio precoz. La edad de comienzo se sitúa entre los 40 y 80 años.
Apolipoproteína E
El gen de ApoE se localiza en el cromosoma 19, y tiene tres pares de alelos posibles: e2, e3, e4. Existe una asociación del genotipo e4 con las formas precoces y tardías de la eA (46), de manera que la herencia de ApoE e4 aumenta el riesgo de desarrollar la enfermedad. La presencia de 1 e4 triplica el riesgo de padecer la enfermedad, y cuando se es portador de 2 e4 el riesgo se incrementa hasta 9 veces. ApoE e4 es un factor de riesgo y no un gen causante porque no todos los portadores del alelo desarrollan el trastorno. El patrón de herencia de la EA de inicio tardío está más asociado a otros trastornos relacionados con la edad (cáncer, aterosclerosis), en los cuales factores ambientales y genéticos desempeñan un rol.
ApoE se une a Ab y la formación del complejo parece estar mediada por oxígeno. ApoE e4 oxidado se une a péptidos Ab más rápidamente que ApoE e3. Esta unión favorece la formación de láminas beta tóxicas y fibrillas de amiloide in vitro. Sin embargo, todavía no se ha elucidado los mecanismos por los cuales ApoE favorece el desarrollo de la EA. Ambas lipoproteínas inhiben la nucleación de la Ab, pero ApoE e3 lo hace con más potencia que ApoE e4.
Alfa-1-antiquimiotripsina (ACT)
Recientes estudios genéticos han demostrado que la posesión del alelo e4 de ApoE no es suficiente por sí solo para explicar la manifestación de la enfermedad de Alzheimer. Se ha comprobado una relación entre los depósitos de b-amiloide, la a1-antiquimotripsina (ACT) y la ApoE que evidencian un involucramiento directo entre el polimorfismo bialélico de la ACT conjugado con la posesión del alelo E4 de ApoE ligado directamente con la aparición de la enfermedad de Alzheimer (67,68,69).
Dihidrolipoil-succinil-transferasa (DSLT)
El gen de la encima DSLT se localiza en el cromosoma 14q24.3. Esta encima es un constituyente fundamental del complejo KGDHC del ciclo de Krebs, y es conocida la existencia de anomalías mitocondriales en la EA; por tanto, una mutación en el gen DSLT constituye un factor de riesgo en personas de edad avanzada con EA.
Existen dos teorías básicas que pretenden explicar el origen de la EA:
TEORÍA b-AMILOIDE
La deposición, a nivel extracelular, del péptido
b-amiloide se encuentra frecuentemente en el cerebro humano envejecido. Inicialmente se forman unas pocas placas en la región basal de la neocorteza. Gradualmente va aumentando el número de depósitos, y finalmente la corteza entera y las partes adyacentes de la sustancia blanca se ven involucradas. Se observa una relación inversa entre el grado de mielinización cortical y la densidad de los depósitos del péptido Ab, el cual se deposita de manera más esparcida en las áreas corticales ricas en mielina que en las zonas pobremente mielinizadas. No hay una relación consistente entre la intensidad de los depósitos de amiloide y la severidad de la disfunción cortical. En los estadios iniciales de EA se observa una alta densidad de ONs y HNs, aunque ninguna parte de las neuronas con ONs están en contacto con depósitos de Ab.La A
b está distribuida en un gradiente de concentración en forma de diana en los cuales las neuronas o sus procesos se localizan en los epicentros (áreas de mayor concentración) de los gradientes (70). También se encuentra inmunoreactividad a lo largo de las membranas neuronales. Estudios sugieren que las neuronas pueden servir como el nido para la génesis de las placas.La angiopatía amiloide ocurre generalmente en estadios más avanzados de la EA y es rara en individuos de mediana edad sin demencia en los cuales se detecta solamente placas corticales difusas.
Considerando que las mutaciones en los genes de PPA o presenilinas no están presentes en ancianos normales (en los cuales la deposición de A
b es muy prevalente) o en casos esporádicos de EA, ¿qué es lo que causa la acumulación de Ab? (1). Para contestar esta pregunta debemos recordar lo que ocurre en la mayoría de las enfermedades genéticas: las mutaciones causan una alteración en la función biológica de la proteína afectada. Por tanto, se puede explicar de manera análoga, la amiloidosis en ausencia de mutaciones de PPA por varios mecanismos que podrían alterar la función biológica de PPA. Estos mecanismos pueden incluir alteraciones de otras proteínas que interaccionen con PPA (p. ej: secretasas, lipoproteínas o chaperonas) o insultos ambientales, tales como la oxidación de PPA o de Ab, o de proteínas que interaccionen con PPA o Ab.Un gran reto en el campo de la PPA sería la identificación de factores que aumentan el procesamiento amiloidogénico de PPA en la EA esporádica. Como ocurre con muchas proteínas alteradas, un metabolismo alterado de PPA podría resultar en una producción más alta de la proteína, conduciendo a un aumento de fragmentos amiloidogénicos.
La producción de amiloide es la consecuencia de un procesamiento anormal de PPA, quizás de forma independiente del nivel de expresión de PPA o de una falta de eliminación de péptidos amiloide producidos normalmente.
TEORÍA TAU
En la EA se produce una acumulación de la proteína tau a nivel intracelular (71). Las células que están en las conexiones ipsilateral-cortical son particularmente susceptibles a desarrollar cambios en el citoesqueleto neuronal. La evolución de estas anormalidades citoesqueléticas se observa mejor en las primeras fases de la EA, y en cerebros de individuos comparativamente más jóvenes (43). Bajo estas condiciones, la patología citoesquelética puede ser estudiada en absencia de depósitos de
b-amiloide y cambios vasculares, o otras lesiones patológicas. Anticuerpos específicos (AT 8) que reaccionan con la proteína tau anormalmente fosforilada, decoran las células piramidales alteradas, y se representa secuencialmente todo el proceso de degeneración neuronal, parecido a las secciones teñidas con la impregnación con plata. Las neuronas AT 8-inmunoreactivas no muestran destrucción de sus neuritas. El hecho de que la proteína tau fosforilada AT 8-positiva aparezca transitoriamente durante ciertas fases de la división celular, hace pensar que las células piramidales no mitóticas AT 8-positivas tienen un control parcial de su ciclo celular y exhiben un inicio abortivo del proceso de división celular. Estas células contienen proteína tau anormal soluble, pero la agregación a ONs o HNs todavía no ha tenido lugar. Los cambios más notables ocurren en el segmento distal de las dendritas, las cuales desarrollan apéndices cortos. Los segmentos dendríticos distales alterados probablemente pierden su conexión a las partes proximales. En este estadio de destrucción empieza la formación de HNs en los segmentos dendríticos alterados y aparición de ONs en el soma. Los primeros rasgos de la existencia de material anormal citoesquelético se observan con una estrecha asociación con depósitos de lipofucsina intraneuronal (72). En algunos tipos de neuronas, los ONs se extienden hasta las dendritas proximales, mientras que en otros tipos permanece confinado al cuerpo celular. Los ONs nunca se extienden al axón proximal. Los cambios en los procesos celulares causan perturbaciones severas en la función neuronal mucho antes de la desaparición final de los cuerpos celulares de las células corticales con ONs. Mueren por apoptosis y necrosis.Las células piramidales normales muestran un núcleo central y material Nissl bien desenrrollado. En las células ovilladas, por el contrario, la capacidad de tinción del material basófilo es menor, y el núcleo se localiza excéntricamente. Después del deterioro del soma celular, el material citoesquelético patológico se convierte en ovillo «fantasma» extraneuronal. Durante este proceso, el ON se hace menos enrroscado y gradualmente pierde su argirofilia específica. Las células piramidales corticales contienen un ovillo durante años. La formación de ovillos «fantasmas» ocurre sólo en áreas y capas de destrucción especialmente temprana y sólo en estados relativamente tardíos de la enfermedad. A medida que el tiempo pasa, incluso los ovillos «fantasmas» desaparecen del tejido. Los granos de lipofucsina de las células de proyección cortical ovilladas pasan a ser extraneuronales, y se puede realizar una tinción específica de estos componentes para indicar la posición inicial de las células nerviosas perdidas. Muchos tipos de neuronas de circuito local son extrordinariamente vulnerables, y desaparecen rápidamente del tejido cortical. Algunas neuronas son sensibles a la desconexión de sus aferentes, mientras que otras muestran una marcada sensibilidad a la hipoxia o influencias nocivas. Las neuronas de circuito local cortical que contienen proteínas de unión al calcio parecen resistir al desarrollo de cambios neurofibrilares.
MUERTE NEURONAL: APOPTOSIS Y NECROSIS
En el tejido con EA se encuentran tanto células apotóticas como necróticas (72,73). Por tanto apoptosis y necrosis deben solapar. Actualmente hay una evidencia considerable de que una mezcla de los dos acontecimientos contribuye a la neurodegeneración en EA y a su patología final.
Apoptosis en la EA
Marcaje apoptótico del DNA in situ.
A
PS1 y PS2 aumentan la sensibilidad apoptótica de las células neuronales.
Colocalización de caspasas y fragmentos de DNA.
Necrosis en la EA
A pesar de que la tinción TUNEL es positiva, no existen características morfológicas de apoptosis in situ.
A
A pesar de que hay un gran número de células con DNA fragmentado en tejido con EA, sólo unas cuantas células muestran características apoptóticas.
ESTRÉS OXIDATIVO
El rol de los radicales libres en el proceso de envejecimiento es un tema de interés actual. Dada la íntima asociación entre envejecimiento y EA, el estrés oxidativo puede tener un papel en la patogenia de las lesiones de EA (74,75). Los radicales libres también están implicados, al menos en parte, en muchas condiciones patológicas asociadas con la edad avanzada como cáncer, enfisema y aterosclerosis.
Peroxidación lipídica, oxidación proteica, fragmentación del DNA nuclear y mitocondrial y formación de productos finales de glicación avanzada en las neuronas de un paciente con EA, indican que su cerebro está sufriendo las consecuencias del estrés oxidativo, que no tardará en exagerar y derivar en neurodegeneración y muerte neuronal.
Una de las evidencias de estrés oxidativo, es la presencia de marcadores oxidativos colocalizados en las lesiones neuropatológicas de la EA (PSs y ONs). La identificación de enzimas antioxidantes (indicadores clave del estrés oxidativo) fue la primera evidencia documentada de la neurotoxicidad oxidativa en las lesiones de la EA. Otros marcadores de estrés oxidativo colocalizados con PSs y ONs son: proteínas del choque térmico, enzimas lisosomales, carbonilos proteicos y peróxidos lipídicos.
El péptido A
b es neurotóxico, conteniendo por tanto, las PSs, una gran capacidad de generación de radicales libres. Uno de los receptores más conocidos del péptido b-amiloide es el RAGE, presente en neuronas y microglía, que es también el receptor de los productos finales de glucosilación avanzada (AGEs). Los AGEs son proteínas modificadas por glucosilación no enzimática que se acumulan con la edad. Se forman cuando la glucosa u otros reactivos carbonilos reaccionan no enzimáticamente con los grupos amino de las proteínas y, después de la oxidación y deshidratación (reacción de Maillard), forman complejos insolubles de proteínas entreunidas covalentemente contribuyendo a la formación de depósitos proteicos intra y extracelulares en la EA, como PSs, ONs y Cuerpos de Hirano. Los AGEs, por otro lado, activan la micro y astroglía, induciendo la formación de citoquinas y radicales libres. Anticuerpos específicos contra dos aductos, piralina y pentosidina permiten localizar AGEs asociados a ONs y PSs. La interacción de Abs y AGEs con RAGEs, en neuronas que expresan este receptor, induce la producción de radicales de oxígeno reactivos (H2O2, radicales hidroxilo, óxido nítrico, anión superóxido, anión peroxinitrito...), y acarrean, como consecuencia, daño neuronal por estrés oxidativo y activación de la cascada inflamatoria, que desencadenarán al final en la muerte neuronal.Una de las consecuencias mejor documentadas del estrés oxidativo, es la oxidación proteica, donde las cadenas laterales son directamente oxidadas para generar modificaciones carbonilo y/o puentes disulfuro cisteína-cisteína. La reacción de los carbonilos con otras cadenas laterales de proteínas, especialmente grupos amino lisina, da como resultado la formación de uniones intra y intermoleculares de proteínas. La modificación del carbonilo también puede ocurrir indirectamente a través de la reacción de metabolitos (que contienen carbonilos) con proteínas. El mejor ejemplo caracterizado de oxidación proteica es el caso de los AGEs.
MODELO PERIFÉRICO DE LA EA: DIAGNÓSTICO «IN VIVO»
El sistema inmunitario de los enfermos de Alzheimer se encuentra alterado, de manera que el análisis de ciertos parámetros (activación macrofágica, incidencia de apoptosis, reactividad frente a insultos oxidativos) puede ser una buena herramienta para el diagnóstico y pronóstico de la EA.
La predisposición genética afecta a todo el individuo, así se puede hablar de una condición Alzheimer que tiene una manifestación más evidente en el cerebro, pero que es concebible encontrar en otros sistemas del cuerpo, particularmente en el sistema inmunitario periférico. La «condición Alzheimer» se manifiesta a través de linfocitos, de manera que los linfocitos Alzheimer se comportan diferente de los linfocitos de individuos comparables pero no afectados (76).
Actualmente se intenta averiguar hasta qué punto el estado fisiopatológico de los linfocitos circulantes puede ser indicativo de una enfermedad neurodegenerativa como el Alzheimer (76,77).
AGRADECIMIENTOS
Parcialmente financiado por FIS 00/0606 y FIS 01/1609 (Ministerio de Sanidad y Seguridad Social) y Union Europea QLK6-CT-1999-02112. X. Gironés es becario del Ministerio de Educación, Cultura y Deportes y A. Guimerá del Departament de Universitats, Recerca i Societat de la Información.
BIBLIOGRAFÍA
Pappolla MA. La Neuropatología y la Biología Molecular de la Enfermedad de Alzheimer. pp. 543-553. En: Neuropathologia. Diagnóstico y Clínica. Cruz-Sánchez FF. Ed. Edimsa. 2000.
Braak H, Braak E. Neuropathological staging of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239-259.
Ferrer I, Cruz-Sánchez FF, Guionnet N, Tuñon T. Estudio de las placas seniles con un método combinado en el cerebro de pacientes con enfermedad de Alzheimer. Arch Neurobiol 6, 53: 222-227, 1990.
Braak H, Braak E, Kalus P. Alzheimer’s disease: A real and laminer pathology in the occipital cortex. Acta Neuropathol, 77: 494-506, 1989.
Ikeda K, Haga C, Kosaka K, Oyanagi S. Senile plaque like structures: Observation of a probably unknown type of senile plaque by periodic-acid methenamine silver (PAM) electron microscopy. Acta Neuropathol, 78: 137-142, 1989.
Yamaguchi H, Hirai S, Morimatsu M, Shoji M, Harigaya Y. Diffuse type of senile plaques in the brains of Alzheimer-type dementia. Acta Neuropathol, 77:113-119, 1988.
Wisniewski HM, Terry RD. Reexamination of the pathogenesis of the senile plaque. En: Zimmerman HM (edit.), Progress in neuropathology. Vol.III, Grune and Stratton, New York and London, 1-26, 1973.
Byrne EJ, Lennox G, Lowe J, Godwin-Austen RB. Diffuse Lewy body disease: clinical features in 15 cases. J Neurol Neurosurg Psychiatry 1989; 52: 709-717.
Selkoe DJ, Bell DS, Podinsy MB, Price DL, Cork LC. Conservation of brain amyloid proteins in aged mammals and humans with Alzheimer’s Disease. Science 1987; 235: (4791) 873-877.
Mann DMA. Diffuse plaques in the cerebellum and corpus striatum in Down’s syndrome contein amyloid b protein only in the form of ab42-43. Neurodegeneration 1996; 5: 115-20.
Dickson DW, Ruan D, Crystal H, Mark HM, Davies P, Kress Y, Yen SH. Hippocampal degeneration differentiates diffuse Lewy body disease (DLBD) from Alzheimer’s Disease: light and electron microscopic immunocytochemistry of CA2-3 neurites specific to DLBD. Neurology 1991, 41: 1402-1409.
Cruz-Sánchez FF. Antigenic determinant properties of neurofibrillary tangles. Relevance to progressive supranuclear palsy. J Neural Transm 42: 165S-178S, 1994.
Cruz-Sánchez FF, Rossi ML, Cardozo A, Tolosa E. Clinical and pathological study of two patients with progressive supranuclear palsy and Alzheimer’s changes. Antigenic determinants that distinguish cortical and subcortical neurofibrillary tangles. Neurosci Lett 136: 43-46, 1992.
Alizadeh-Khiavi K, Normand J, Chronopoulos S, Ali-kan Z. Alzheimer’s disease brain derived ubiquitin has amyloid-enhancing factor activity: behavior of ubiquitin during accelerated amyloidogenesis. Acta Neuropathol 1991; (Berl) 81: 280-286.
Lowe J, Mayer RJ, Landon M. Ubiquitin in neurodegenerative diseases. Brain Pathol 1993; 3: 55-65.
Cruz-Sánchez FF, Marin C, Rossi ML, Cardozo A, Ferrer I, Tolosa E. Ubiquitin in cerebral amiloid angiopathy. J Neurological Sciences 112: 46-50, 1992.
Braak H, Braak E. Neuropil threads occur in dendrites of tangle-bearing nerve cells. Neuropathol Appl Neurobiol 1988; 14: 39-44.
Probst A, Anderton BH, Brion JP, Ulrich J. Senile plaque neurites fail to demonstrate anti-paired helical filaments and microtubule associated protein tau immunoreactive proteins in absence of neurofibrillary tangles of the cortex. Acta Neuropathol 1989; 77: 430-436.
Hauw JJ, Verny M, Delaère P, He Y, Duyckaerts C. Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer’s disease and aging. Neurosci Lett 1990; 119: 182-186.
Yamaguchi H, Nakazato Y, Shoji M, Ihara Y, Hirai S. Ultrastrucrure of the neuropil threads in the Alzheimer’s brain: the dendritic origin and accumulation in the senile plaques. Acta Neuropathol 1990; 80: 368-374.
Perry G, Kawai M, Tabaton M, et al. Neuropil threads of Alzheimer’s disease show a marked alteration of the normal cytoskeleton. J Neurosci 1991; 11: 1748-1755.
Cervós-Navarro J, Urich H. Metabolic and degenerative diseases of the central nervous system. Ed.Academic Press, 1995.
Okamoto K, Hirai S, Iizuka T, Yanagasina T, Watanabe M. Reexamination of granovacuolar degeneration. Acta Neuropathol 1991; 82: 340-345.
Tomonaga M, Yamanouchi H, Kameyama M. Hirano bodies observed in brain of the aged. Jpn J Geriatr 1975; 12: 13-17.
Mann DMA. The neuropathology of Alzheimer’s disease: a review with pathogenic, aetiological and therapeutical considerations. Mech Aging Dev 1985; 31: 213-255.
Cervós-Navarro J. Modifications in the genetic expression in the aging brain. En: Wertheimer J, Marois M (eds). Senile dementia: outlook for the future. Liss New York, 1984; pp 113-123.
Wood PL, Etienne P, Lal S, Nair NPV, Finlayson MH, Gauthier S, Palo J, Haltia M, Pateau A, Bird ED. A post-mortem comparison of the cortical cholinergic system in Alzheimer’s disease and Pick’s disease. Neurol Sci 1983; 62: 211-216.
D’Amato RJ, Zweig RM, Whitehouse PJ, Wenk GL, Singer HS, Mayeux R, Price DL, Snyder SH. Aminergic systems in Alzheimer’s disease. Ann Neurol 1987; 22: 229-236.
Cowburn RJ, Barton ATL, Hardy JA, Wester P, Winblad B. Region-specific defects in glutamate and gamma-aminobutyric acid innervation in Alzheimer’s disease. Biochem Soc Trans 1987; 15: 505-506.
Muñoz-García DG, Wang D, Greenberg BD. Hirano bodies accumulate c-terminal sequences of b-amyloid precursor protein (b-APP) epitopes. J Neuropathol Exp Neurol 1993; 52: 14-21.
Río-Hortega P Del. El tercer elemento de los centros nerviosos II. Intervención de la microglía en procesos patológicos. Bol de la Soc Esp de Biol 1920; 8: 91-103.
Piani D, Frei K, Do KQ, Cuenod M, Fontana A. Murine brain macrophages induce NMDA receptor mediated neurotoxicity in vitro by secreting glutamate. Neurosci Lett 1991; 133: 159-62.
Nakajima M, Kohsaka S. Functional implications of microglia-derived secretory proteases. En: Ling EA, Tan CK, Tan CBC,eds. Topical Issues in microglial research. Singapore: Singapore Neuroscience Association. 1996: 203-18.
Moser M. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Washington, DC, American Psychiatric Association. 1994.
Mckhann G, Drachman D, Folstein M, et al. Clinical Diagnosis of Alzheimer’s Disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurol 1984; 34: 939-944.
Tierney MC, Fisher RH, Lewis AJ, Zorzitto ML, Snow WG, Reid DW, Nieuwstraten P. The NINCDS-ADRDA Work Group Criteria for the Clinical diagnosis of probable Alzheimer’s Disease: a clinicopathologic study of 57 cases. Neurology 1988; 38: 359-364.
American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 3rd ed. Revised. Washington DC: Am Psychia Assoc. 1987.
Duque-Castaño A, Roldán ML, Arando-Viana JC, Arcos-Burgos M, Cubillo H, Lopera F. Hallazgos neuropatológicos en la enfermedad de Alzheimer de inicio temprano (mutación E280A-PS1). Rev Neurol 1999; 29(1): 1-6.
Lopera F, Tobón N, Arcos-Burgos M, Vargas S, Gutiérrez JE, Rosselli M, Ardilla A. Caracterización imagenológica de la enfermedad de Alzheimer asociada a la mutación E280A-PS1. Estudio caso-control: hallazgos en la resonancia magnética. Rev Neurol 1999; 29: 6-12.
Morris C, Heyman JA, Mohs RC, Hughes JP, Van Belle G, Fillenbaum G, Mellits ED, Clark C. The Consortium to establish a registry for Alzheimer’s disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 1989; 39: 1159-1165.
Mirra SS, Heyman A, Mckeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, Van Belle G, Berg L. The Consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991; 41: 479-486.
Khachaturian ZS. Diagnosis of Alzheimer’s Disease. Arch Neurol 42 (1985) 1097-1105.
Braak Heiko and Braak Eva. Cortical destruction and cell death in Alzheimer’s disease. pp. 497-507. En: Koliatsos, Ratan. Cell death and diseases of the nervous system. Ed. Humana Press.1999.
Everall IP, Deteresa R, Terry R. Comparison of two quantitative methods for the evaluation of neural number in the frontal cortex in Alzheimer’s Disease. J Neuropathol Exp Neurol 1997; 56: 1202.
Wiesniewski HM, Merz GS. Neuropathology of the aging brains and dementia of Alzheimer type. En: Gaitz , Samorajski (Eds.), Aging 2000: Our Health Care Density, Vol.1, Springer, New York, 1985, pp. 231-243.
Cruz-Sánchez FF, Durany N, Thome J, Riederer P, Zambón D. Correlation between apolipoprotein E polimorphism and Alzheimer’s disease pathology. Journal of Alzheimer’s Disease 2 (2000) 223-229. IOS Press.
Bogdanovic N, Morris JH. Diagnostic criteria for Alzheimer’s Disease in multi-centre Brain banking. En: Cruz-Sánchez FF y cols. Neuropathological Diagnostic Criteria for Brain banking. Ed. IOS Press. 1995.
Ferrer I, Guionnet N, Cruz-Sánchez FF, Tuñon T. Neuronal alterations in patients with dementia: a golgi study on biopsy samples. Neurosci Lett 114: 11-16, 1990.
Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and parkinsonism linked to chromosome 17: A new group of tauopathies. Brain Pathol 1998; 8: 387-402.
Pick A. Über die Beziehungen der senile Hirnatropie zur Aphasie. Prag Med Wschr 1982; 17: 165-167.
Smith DA, Lantos PL. A case of combined Pick’s disease and Alzheimer’s disease. J Neurol Neurosurg Psychiatry 1983; 46: 675-677.
Hof PR, Bouras C, Perl DC, Morrison JH. Quantitative neuropathologic analysis of Pick’s disease cases: cortical distribution of Pick bodies and coexistence with Alzheimer’s disease. Acta Neuropathol 1994; 87: 115-124.
Lowe J, Lennon G, Jefferson D, Morrell K, McQuire D, Gray T, Ladon M, Doherty FJ, Mayer RJ. A filamentous inclusion body within the anterior horn neurons in motor neuron disease defined by immunocytochemical locations of ubiquitin. Neurosci Lett 1988; 94: 203-210.
Terry RD, Hansen LA, De Teresa R, Davies P, Tobias H, Katzman R. Senile dementia of the Alzheimer type without neocortical fibrillary tangles. J Neuropathol Exp Neurol 1987; 46: 262-268.
Hansen LA, Masliah E, Terry RD, Mirra SS. A Neuropathological subset of Alzheimer’s disease with concominant Lewy body disease and spongiform change. Acta Neuropathol 1989; 78: 194-201.
Braak H, Braak E. Cortical and subcortical argyrophilic grains characterise a disease associated with adult onset dementia. Neuropathol Appl Neurobiol 1989; 15: 13-26.
Mizutani Y, Yokochi M, Oyanagi S. Juvenile parkinsonism: a case with first clinical manifestation at the age of six years and with neuropathological findings suggesting a new pathogenesis. Clin Neuropathol 1991; 10: 91-97.
Gil Nécija Eulogio. Diagnóstico biológico. Cuarto curso nacional de Enfermedad de Alzheimer. Sevilla, 23-24 septiembre 1999. Ed. Andrómico.
Farrer L, O’Sullivan DM, Cupples LA, Growdon JH, Myers RH. Assesment of genetic risk for Alzheimer’s disease among first degree relatives. Ann Neurol 1989; 25: 485-493.
Huff J, Auerbach J, Chakravarti A, Boller F. Risk of dementia in relatives of patients with Alzheimer’s disease. Neurol 1988; 30: 786-790.
Miller DL, Papayannopoulos IA, Styles J, Bobin SA, Lin YY, Biermann K, Iqbal K. Peptide compositions of the cerebrovascular and senile plaque core amyloid deposits of Alzheimer’s disease. Arch Biochem 1993; B301: 41-52.
Masters Cl, Simms G, Weinman NA, Multhaup G, Mc Donald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer’s disease and Down syndrome. Proc Natl Acad Sci USA 1985; 82: 4245-4249.
Kennedy AM, Newman S, Mc Caddon A, Ball J, Roques P, Mullar M, Hardy J, Chartier-Harlin MC, Frackowiak RS, Warrington EK. Familial Alzheimer’s disease. A pedigree with a miss-sense mutation in the amyloid precursor protein gene. Brain 1993; 116: 09-324.
Driscoll M, Chalfie M. The mec-4 gene is a member of a family of Caenorhabditis elegans genes that can mutate to induce neuronal degenaeration. Nature 1991; 349: 588-593.
Tagliavini F, Giaccone G, Frangione B, Bugiani O. Preamyloid deposits in the cerebral cortex of patients with Alzheimer’s disease and nondemented individuals. Neurosci Lett 1988; 93: 191-195.
Yanker Da, Duffy LK, Kirschner Da. Neurotrophic and neurotoxic effects of amyloid b protein: reversal by tachykin neuropeptides. Science 1990; 250: 279-282.
Durany N, Ravid R, Riederer P, Cruz-Sánchez FF. Increased frequency of the alpha-1-antichymotrypsin T allele in cerebral amyloid angiopathy. Neuropathology 2000; 20, 184-189.
Durany N, Morgan K, Thome J, Tilley L, Kalsheker N, Münch G, Riederer P, Cruz-Sánchez FF. Relationship between alpha-1-antichymotrypsin micro-satellite and apolipoprotein E in Alzheimer’s disease. Alzheimer’s Report 1 (5): 315-320, 1998.
Durany N, Cruz-Sánchez FF, Thome J, Höhn H, Retz W, Riederer P, Rösler M. No association between alpha-1-antichymotrypsin and apolipoprotein E in Alzheimer’s disease and vascular dementia. Alzheimer’s Report 2: 159-164, 1999.
Pappolla MA, Omar RA, Sambamurti K, Anderson J, Kim KS, Robakis NK. The genesis of the senile plaque II. Further evidence in support of its neural origin. Am J Pathol 1992; 141: 1151-1159.
Ribalta T, Rey MJ, Blesa R, Tolosa E, Cardesa A, Ferrer I. Distribution of Tau pathology in the brain stem of patiens with Alzheimer’s disease. Brain Pathology. Vol 10, No 4; pp. 514. Sept. 2000.
Cruz-Sánchez FF, Cusidó M, Ruiz-Ávila L, Esquerda J. Patología neuronal. Pp. 21-56. En: Cruz-Sánchez FF, Neuropathologia.Diagnóstico y clínica. Ed. Edimsa. 2000.
Behl C. Apoptosis and Alzheimer’s disease. Journal of Neural Transmission (2000) 107: 1325-1344.
Flint Beal M. Role of mitochondria and oxidative damage in Alzheimer’s disease. pp. 89-93. En: Flint Beal M. Mitochondrial disfunction and oxidative damage in neurodegenerative diseases. Ed.Springer-Verlag. 1995.
Smith Mark A, Sayre Lawrence M, Perry G. Morfhological aspects of oxidative damage in Alzheimer’s disease. pp. 335-340. En: Beal, Howell, Bodis-Wollner. Mitochondria and free radicals in neurodegenerative diseases. Ed.Wiley-Liss. 1997.
Hartwig M, Schulz J, Ferszt R, Kühl K-P, Cervós-Navarro J. Chronic activation of peripheral blood macrophages in Alzheimer’s Disease. Alzheimer’s Reports 1999; 2, 225-229.
Gironès X, Chyan YJ, Ariza A, Riutort N, Fernández MA, Cervós-Navarro J, Latorre P, Cruz-Sánchez FF. Macrophage activation and celldeath in Alzheimer disease: Effects of age and apolipoprotein E genotype. Neuropathol Appl Neurobiol (submitted).
![]()